








en los últimos años es cada vez más frecuente enfrentarse a nuevos fármacos o nuevas indicaciones de viejos medicamentos que surgen de estudios de equivalencia. En particular los denominados estudios de no-inferioridad (ENI).
Objetivocomentar y analizar los aspectos metodológicos y éticos de los ENI, en particular en Neurología.
Desarrolloa través de una revisión narrativa se analiza la terminología para este tipo de estudios, las situaciones en las que se puede considerar un ENI, el tamaño de la muestra e hipótesis planteada y sus diferencias respecto a un estudio convencional de superioridad. También los factores que influyen en la elección del margen de no inferioridad, tanto los estadísticos como los clínicos, y finalmente los aspectos regulatorios y éticos.
Conclusioneslos ENI son cada vez más frecuentes en el ámbito de la Neurología y presentan aspectos metodológicos y éticos particulares, que deben ser conocidos por el neurólogo para poder interpretar adecuadamente el real beneficio de una nueva terapia.
In recent years, it has become more frequent to find new treatments or new prescriptions of old drugs that arise from equivalence trials, particularly the so called non-inferiority trials (NITs).
ObjectiveTo comment on and analyze the methodological and ethical aspects of NITs in neurology.
FindingsThis narrative review analyzes the terminology for this type of studies, the situations in which an NIT can be considered, the sample size, and the hypothesis set up as well as the differences between an NIT and a conventional superiority trial. The review includes the discussion of factors, both statistic and clinical ones, that influence the choice of the non-inferiority margin, and of regulatory and ethics issues.
ConclusionsNon-inferiority trials are becoming more frequent in the setting of neurology. They present specific methodological and ethical aspects that should be considered by the neurologist in order to adequately interpret the real benefit of a new therapy.
En los últimos años es cada vez más frecuente enfrentarse a nuevas intervenciones (fundamentalmente relacionadas con fármacos) o nuevas indicaciones de viejos medicamentos que no surgen sólo de ensayos de superioridad, sino de estudios de equivalencia, en particular ensayos denominados de noinferioridad. En estos se compara una nueva terapia con un control activo que habitualmente es un tratamiento estándar para el trastorno que se evalúa.
A modo de representación, en la actualidad se encuentran registrados 248 ensayos clínicos de no-inferioridad de fase III, sólo en el registro del National Institutes of Health (NIH), de los cuales 62 se hallan reclutando pacientes. Al menos unos 30 involucran enfermedades neurológicas1.
¿Equivalencia?Genéricamente el término equivalencia se ha utilizado en diferentes situaciones experimentales de forma algo confusa. Un estudio, por ejemplo, demostró que sólo el 3% de los trabajos publicados como de equivalencia lo eran realmente2, por lo que es conveniente aclarar primero a qué nos referimos.
Últimamente equivalencia se ha utilizado para englobar dos formas diferentes de estudios de investigación: estudios de equivalencia y de no-inferioridad.
Las guías incluidas en la International Conference of Harmonization (ICH) definen un estudio de equivalencia como aquel cuyo objetivo primario es demostrar que dos o más tratamientos difieren en una magnitud que no tiene relevancia clínica, estableciendo que la diferencia real se encuentra entre un margen (de equivalencia) superior e inferior preestablecidos3, es decir, que la equivalencia está definida como el efecto del tratamiento que está entre un delta inferior (−δ) y uno superior (δ)4.
Un verdadero estudio de equivalencia (2-colas) no es habitual, ya que la mayoría de los estudios se plantean la no-inferioridad, es decir, que no sea peor que el tratamiento convencional4. Es por esto que se prefiere no utilizar actualmente el concepto de equivalencia: si la nueva estrategia es superior a la previa, no es equivalente5.
Recordemos que en un estudio de superioridad se evalúa si un tratamiento es mejor que un control, ya sea que se trate de otra estrategia de tratamiento o de un placebo para un desenlace dado. Por lo tanto, si no se demuestra superioridad nuestra intención principal es establecer que un nuevo tratamiento no sea peor (inferior) que una terapia estándar aceptada.
Un estudio de no-inferioridad, en cambio, puede ser definido como aquel que busca determinar si un nuevo tratamiento “no es peor” que un tratamiento de referencia. Debido a que probar igualdad es imposible (se requeriría de una muestra infinita) se establece un margen de no-inferioridad (δ) para el efecto del tratamiento para un desenlace dado4.
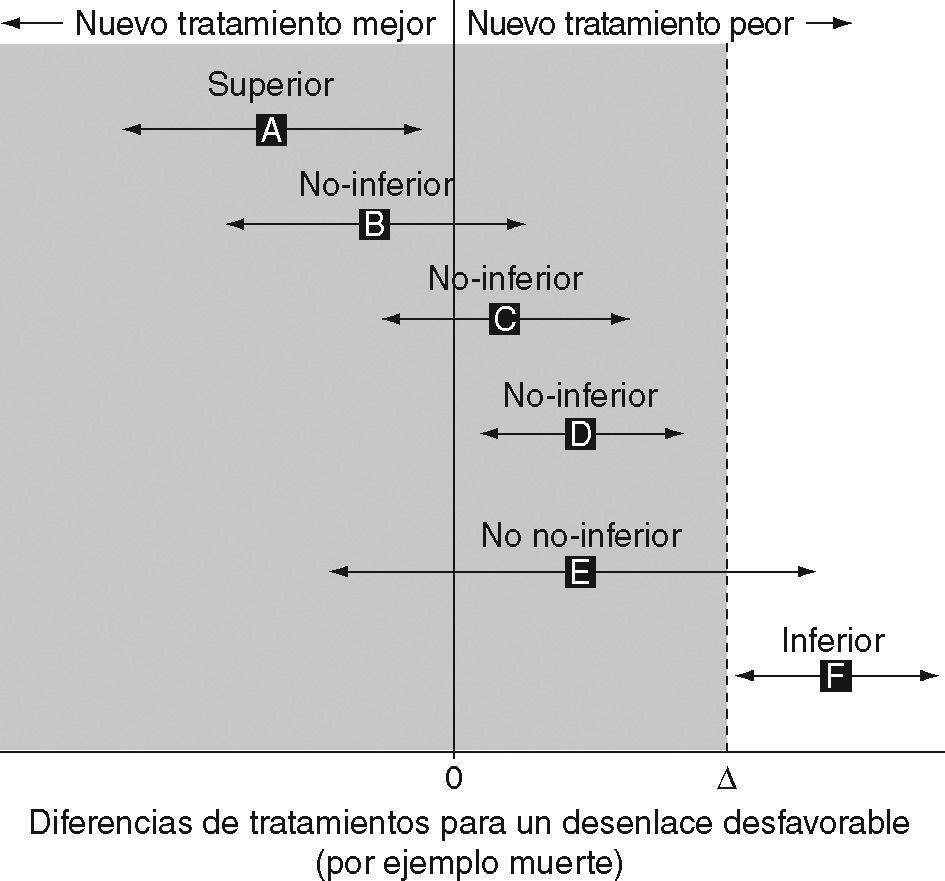
¿Cuáles son entonces los aspectos conceptuales básicos de un estudio de no-inferioridad?: comparación con otro tratamiento activo (no placebo) y expectativa de que el efecto mínimo del nuevo tratamiento no sea menor al efecto establecido para el tratamiento estándar o convencional. Dicho de otra manera, intentan demostrar que una nueva intervención tiene al menos tanto efecto como la estándar, o que incluso el nuevo tratamiento puede ser peor hasta un delta tolerable y preestablecido, bajo la promesa de algún beneficio extra (fig. 1).
 del 95%. Línea 0: representa el efecto del control activo. En la situación A, dado que el IC inferior supera al efecto del control activo, el nuevo tratamiento es superior aunque un estudio de noinferioridad (1 cola) no posee la capacidad para detectarlo. B, C y D representan diferentes situaciones de noinferioridad, el IC inferior del nuevo tratamiento está “por encima” del margen especificado (ver más abajo). En E el tratamiento propuesto es no no-inferior, aunque un IC menor podría hacer que “entre” completamente en el área de no-inferioridad. F: el tratamiento está totalmente por debajo del margen, por lo que es inferior. Modificada de Piaggio G, et al4.")
Línea interrumpida: margen de no-inferioridad. Área sombreada: zona de no-inferioridad. Flechas: intervalos de confianza (IC) del 95%. Línea 0: representa el efecto del control activo. En la situación A, dado que el IC inferior supera al efecto del control activo, el nuevo tratamiento es superior aunque un estudio de noinferioridad (1 cola) no posee la capacidad para detectarlo. B, C y D representan diferentes situaciones de noinferioridad, el IC inferior del nuevo tratamiento está “por encima” del margen especificado (ver más abajo). En E el tratamiento propuesto es no no-inferior, aunque un IC menor podría hacer que “entre” completamente en el área de no-inferioridad. F: el tratamiento está totalmente por debajo del margen, por lo que es inferior. Modificada de Piaggio G, et al4.
Otra forma de pensar un estudio de no-inferioridad es no a través de su metodología, sino a partir de su finalidad como estudio de “suficiencia” (Sufficiency Trial): está diseñado para demostrar que la actividad del fármaco en estudio es, comparada con el control activo, suficiente para cumplir con el propósito del estudio (por ejemplo para apoyar la recomendación de expertos o aprobación regulatoria)6.
Cuándo se puede considerar un estudio de no-inferioridadEn situaciones en las que no es ético involucrar a pacientes en una rama placebo para un trastorno que posee un tratamiento reconocido, es imprescindible comparar nuestra nueva intervención con un control activo. En este caso puede no ser necesario demostrar superioridad, sino tan sólo establecer que la nueva terapia funciona tan bien como la estándar, con ciertas ventajas como perfil de menor toxicidad y efectos adversos7, menor coste, o ser menos invasivo4. En enfermedades graves, por ejemplo, se puede contemplar una pequeña disminución en la sobrevida si la nueva intervención ofrece una importante mejoría en la calidad de vida. Entonces una nueva estrategia puede estudiarse bajo no-inferioridad si ofrece una misma eficacia que un tratamiento ya establecido, pero con una ventaja en otro desenlace relevante y no considerado previamente8.
Un último aspecto, no menor, es que los ensayos de no-inferioridad reducen además los tiempos y costes de una investigación clínica.
Tamaño de la muestra e hipótesisLa determinación del tamaño de la muestra en un estudio de no-inferioridad no escapa a las reglas generales de un estudio de superioridad: cuanto menor es la diferencia que se espera encontrar, mayor es el tamaño de la muestra necesario y viceversa (proporcional a 1/δ2); así, cuanto más amplio es el margen de no-inferioridad (piso más bajo) menor es el tamaño de la muestra9. Sin embargo, el tamaño de la muestra es menor en estudios de no-inferioridad que en los de superioridad, ya que sólo se necesita verificar que el nuevo tratamiento no es peor, más aún, cuanto mayor es el potencial beneficio de la nueva terapia más se reduce el tamaño de la muestra10.
Las hipótesis que subyacen a un estudio de no-inferioridad también difieren de las de un estudio de superioridad. En este último se parte de la hipótesis nula de no diferencia para demostrar la hipótesis alternativa de diferencia: tratamiento nuevo superior al control. En cambio, en el ámbito de la noinferioridad partimos de la hipótesis nula de que nuestro nuevo tratamiento a probar es inferior (la diferencia absoluta es mayor —excede— al delta especificado), para llegar a la no diferencia (establece que la diferencia absoluta es menor al delta especificado).
Teniendo en cuenta esto, un error tipo I nos llevaría, en un estudio de no-inferioridad, a concluir falsamente que el nuevo tratamiento es no-inferior al estándar, y un error de tipo II a concluir falsamente que el nuevo tratamiento es inferior al estándar8.
Esto nos lleva a considerar otra distinción importante: habitualmente los ensayos de superioridad hacen análisis por “intención de tratar” (según como fueron aleatorizados los pacientes, independientemente de si posteriormente hubo abandono o cambio a otra rama de tratamiento para preservar el efecto de la aleatorización) y “por protocolo” (también llamado “en tratamiento”, según qué tratamiento realmente recibió cada paciente, independientemente de a qué grupo fue aleatorizado). Si un estudio de superioridad que compara dos fármacos presenta un porcentaje elevado de abandono (por efectos adversos de uno de los tratamientos, por ejemplo) en la rama control, ambos tipos de análisis van a diferir sustancialmente en sus resultados; el escenario “en tratamiento” acentúa más las diferencias entre las dos terapias que se estudian, favoreciendo el rechazo de la hipótesis nula de no diferencia, y el análisis de “intención de tratar” tiende a llevar los resultados hacia la hipótesis nula (no diferencia). Precisamente la inversa ocurre en la no-inferioridad; se buscan “similitudes” (hipótesis alternativa de no diferencia) entre los tratamientos. El análisis de “intención de tratar” tiende a llevar los resultados hacia la hipótesis alternativa (no diferencia), favoreciendo a la nueva intervención9. Por ejemplo, en el estudio SPACE se ilustra muy bien la diferencia entre uno y otro tipo de análisis. Allí se comparó la colocación de stent carotídeo frente a endarterectomía carotídea en pacientes con estenosis carotídea sintomática grave. Se había establecido como margen de no-inferioridad que la diferencia absoluta del peor efecto en la rama stent, para el desenlace combinado stroke ipsilateral o muerte por cualquier causa a los 30 días, no estuviera mas allá del 2,5% del mismo desenlace para la rama endarterectomía. Si bien en el estudio SPACE no se demostró la no-inferioridad del stent sobre la endarterectomía, en el análisis por “intención de tratar” este peor efecto estuvo a 0,41% del margen, mientras que en análisis “por protocolo” al 1,26%. Se observa entonces cuán diferentes podrían ser los resultados en ambas situaciones de análisis11,12 (fig. 2).
Establecer el margen de no-inferioridad y superior (+). Debido a que se evalúa un desenlace desfavorable el área de no-inferioridad está por debajo del margen.")
Sin duda hay acuerdo completo en que el margen de no-inferioridad debe haber sido establecido antes de conducir el ensayo, como alguien ha escrito: “correr el margen de no-inferioridad después de conocer los resultados es como apostar a los caballos tras haber visto la carrera”.
Como destacamos anteriormente las guías ICH (ICH E10 1.5.1.1)1 hacen hincapié en la relevancia clínica de la no diferencia entre dos tratamientos para establecer la no-inferioridad de una terapia, sin embargo casi nunca este es el factor determinante del valor de dicho margen9. Esto ha sido destacado particularmente en cáncer8 y epilepsia13. Casi invariablemente son los aspectos estadísticos los que determinan la elección del valor del margen de no-inferioridad.
Dado que cuanto más bajo o más alto sea el piso de eficacia de un tratamiento estándar (control), menor o mayor respectivamente será el salto que tenga que dar el nuevo tratamiento para alcanzar al control, es decir, demostrar que es no-inferior. Resulta evidente entonces que la determinación del margen constituye un aspecto fundamental en la realización de este tipo de estudios.
Dentro de los factores no estrictamente estadísticos que se deben tener en cuenta para establecer el margen de no-inferioridad hay que considerar la gravedad del trastorno que se estudia, la preocupación por la potencial toxicidad del tratamiento y el desenlace primario elegido (si la enfermedad es grave y el desenlace es, por ejemplo, mortalidad se recomienda un delta pequeño)2,8.
Volviendo a los aspectos estadísticos la primera condición estadística para establecer un margen sería que al menos el efecto mínimo del nuevo tratamiento no sea menor al efecto mínimo demostrado para el tratamiento estándar.
Este piso de efecto mínimo demostrado para el tratamiento estándar es uno de los factores que determina el margen de no-inferioridad, pero exige que idealmente se disponga de una medida de efecto con precisión adecuada (intervalos de confianza pequeños), que provenga de estudios previos de alta calidad metodológica (ensayos clínicos aleatorizados y controlados) o metaanálisis con la menor heterogeneidad posible, los cuales no siempre están disponibles. No es posible encarar una comparación con este tipo de diseño si la evidencia de la eficacia del tratamiento estándar proviene de estudios pequeños, de baja calidad metodológica, y como veremos a continuación si sus estudios pivotales o los que se utilizaron para determinar su eficacia para el desenlace que se quiere evaluar han sido realizados mucho tiempo atrás, los pacientes participantes en los mismos difieren de los actuales o si se consideran otros desenlaces. Las guías ICH explican claramente que un ensayo clínico debe tener la “capacidad para distinguir entre terapéuticas activas e inactivas. Si un tratamiento no es superior de forma consistente en ensayos previos, consideraremos el diseño de no-inferioridad inapropiado, porque no podemos presumir que el nuevo estudio tendrá la sensibilidad suficiente” (assay sensitivity).
Tiempo de los ensayos previosPlanteemos la siguiente situación hipotética: tenemos un fármaco A, con demostrada superioridad respecto al placebo en, como mínimo, un 2% (incremento del beneficio absoluto del intervalo de confianza inferior de A frente a placebo) para la enfermedad X. El fármaco A es ahora el tratamiento estándar para X. Diez años después el medicamento B es propuesto para un estudio de no-inferioridad contra A (control activo), con un margen de no-inferioridad del 2% (su efecto mínimo se espera que sea al menos igual que el demostrado para A respecto del placebo).
Pero actualmente, mejoras en el tratamiento de X hacen que si se hiciera un estudio de A contra placebo la diferencia sería mayor que el 2%. Esto puede ocurrir por ejemplo porque A se combina con otras intervenciones farmacológicas o no (sistema de salud, diagnóstico precoz) que potencian su efecto y que no estaban disponibles previamente, pero que ahora son de uso corriente. Esta potenciación sólo afecta al control activo, por lo que el margen debería estar “más alto”, debería ser más exigente. Dado las mejoras en el tratamiento de X desde la introducción de A, B arranca con ventaja en el estudio de noinferioridad, ya que el efecto real de A está subestimado.
Este fenómeno se denomina efecto de dilución de la población, y es un buen ejemplo de un factor no estadístico a considerar cuando se discute el margen de no-inferioridad.
Una forma práctica de minimizar este factor es utilizando medidas de efecto con razones (ratios), en lugar de diferencias absolutas, ya que las medidas relativas permiten una mejor comparación indirecta entre poblaciones y tiempos diferentes, con una misma intervención y desenlace (fig. 3).
Tipo de pacientes, intervención y desenlaces
Los pacientes deben ser lo más semejantes posible respecto del riesgo basal, tipo de enfermedad, variables demográficas y tratamientos concomitantes, así como en la duración del estudio, el desenlace medido, la similitud en el diseño (criterios de inclusión, exclusión, periodos basales o run in, régimen de visitas, etc.), y como vimos antes se debe asegurar la constancia del efecto del tratamiento. Por este motivo los estudios de no-inferioridad, al tener que ajustarse a lo realizado en estudios anteriores con el control activo, requieren de una rigurosidad especial14,15.
Cuán no-inferior es nuestro nuevo tratamiento no-inferiorComo se observa en la figura 1 los tratamientos B, C y D son no-inferiores (su efecto mínimo está por encima del margen preespecificado), pero difieren en su nivel de no-inferioridad; es evidente que el tratamiento D es menos no-inferior que el C y B respectivamente, es decir, que no basta con determinar que nuestro nuevo tratamiento es no-inferior, sino que es necesario saber en qué grado es no inferior.
Una cuestión implícita en el diseño de no-inferioridad es que si el estudio resulta positivo, es decir, se afirma que el nuevo tratamiento B no es inferior al control activo A, puede deducirse en forma indirecta que el tratamiento B superaría al placebo en caso de que ese estudio se efectuara5.
En los ensayos de no-inferioridad los resultados pueden interpretarse de dos maneras: ambos son igualmente eficaces, pero también son igualmente inútiles11.
Esto constituye una paradoja conocida como el problema de la “imputación al placebo”: un estudio de no-inferioridad no demuestra eficacia por sí mismo, sino a través de su sensibilidad (assay sensitivity). Se asume que cuantitativamente el nuevo tratamiento conserva una porción (mayor o menor como en B, C y D) de la superioridad de la terapia estándar sobre el placebo.
Una medida adecuada para medir ese grado de no-inferioridad es la llamada fracción preservada (ƒ): cuánto efecto conserva el nuevo tratamiento del efecto del tratamiento estándar sobre el placebo. En un artículo en el que se realizó un nuevo análisis de los datos de varios estudios de enfermedad cardiovascular y se calculó la fracción preservada para cada uno, se observó que en 5 estudios el límite inferior de la fracción preservada era menor del 50%, incluso cuando tres de ellos habían llegado a la conclusión de no-inferioridad al superar el margen preespecificado15,16. No hay un acuerdo completo en cuál debe ser la ƒ, ni cuál es la mejor forma de establecer el margen de no-inferioridad, pero ningún estudio debería establecer un margen que no considere conservar, como mínimo, la mitad (ƒ) del efecto del control activo9,11.
Es cada vez más frecuente que se utilice la fracción preservada para establecer el margen de no-inferioridad, teniendo este la ventaja de que garantiza un efecto mínimo del nuevo fármaco respecto de su comparación indirecta con el placebo.
En el estudio PRoFESS17, por ejemplo, se comparó en la rama antiagregante la combinación de dipiridamol-aspirina de liberación extendida (que había demostrado un beneficio absoluto del 3% respecto a aspirina en el estudio ESPS-218,19) con clopidogrel para determinar si el primero era no-inferior a clopidogrel. Se estableció el margen con el criterio de mantener al menos el 50% del efecto (fracción preservada) de clopidogrel sobre aspirina, teniendo en cuenta el desenlace de stroke no-fatal del estudio CAPRIE20.
Si consideramos que el beneficio de clopidogrel sobre aspirina en el estudio CAPRIE sólo se observó en el desenlace combinado de stroke isquémico, infarto de miocardio y muerte de causa vascular de las tres cohortes (stroke, infarto de miocardio y arteriopatía periférica), y que no hubo diferencias estadísticamente significativas para ningún desenlace en la cohorte de stroke isquémico tomada como subgrupo20,21, es razonable pensar que preservar la mitad del efecto de un beneficio marginal y no estadísticamente significativo es un buen piso para que otro tratamiento demuestre no ser peor9.
El estudio finalmente falló en demostrar que la combinación dipiridamol-aspirina de liberación extendida era noinferior a clopidogrel, pero es curioso que los autores escriban en la discusión que dicho margen era exigente, atribuyéndole parte de la culpa de la no demostración de no-inferioridad.
A pesar de que fue un estudio bien diseñado y conducido, con un número de pacientes extraordinario y un buen seguimiento, en el estudio PRoFESS algo funcionó mal (probablemente por sobreestimación del efecto de dipiridamol-aspirina en el estudio ESPS-2, eficacia mayor de clopidogrel que la establecida en el estudio CAPRIE y lacunarización de los eventos con reducción del riesgo de recurrencia22), pero seguro que no fue el margen de no-inferioridad.
Analizando con mas detalle el estudio SPACE, si se hace el cálculo de la ƒ con el método de Hasselblad y Kong para Odds ratio23, asumiendo la tasa de eventos y el número de pacientes originalmente planteado en el protocolo para demostrar la no-inferioridad (es decir, haciendo el cálculo hipotético como si hubiera demostrado la no-inferioridad del stent frente a endarterectomía), vemos que la ƒ del stent carotídeo es de 1,04 (IC 95%: 0,30 a 1,78). Esto significa que si se hubiera demostrado la no-inferioridad su fracción preservada mínima hubiera sido del 30%, por debajo del mínimo del 50% recomendado habitualmente por la Food and Drug Administration (FDA) para estudios en el área cardiovascular.
En epilepsia hay recomendaciones para establecer el margen de no-inferioridad24, sin embargo ningún ensayo de este tipo en epilepsia publicado hasta la fecha ha utilizado como margen la menor diferencia con relevancia clínica11. En algunos otros estudios la justificación del margen elegido no está clara25,26. En otro estudio se utilizó un margen amplio, a partir de definir un valor de placebo potencial (putative placebo); se trató de un estudio aleatorizado, doble ciego, de dos ramas para demostrar si la monoterapia con levetiracetam (1.000 a 3.000mg/ día) era no-inferior a monoterapia con carbamacepina de liberación controlada (CBZ-CR 400 a 1.200mg/día) en adultos mayores de 16 años con epilepsia de reciente comienzo, con crisis parciales o tónico-clónicas generalizadas. Se estableció el margen de no-inferioridad utilizando como evidencia previa un único estudio identificado como placebo-controlado en pacientes no tratados, que demostró una tasa libre de crisis a los tres meses del 11,5% para la rama placebo. Se estableció entonces una tasa libre de crisis para placebo “conservadora” de un 15%. Se dividió a la mitad la diferencia entre CBZ-CR esperada (45%) y la estimada para placebo (15%)27, es decir, efecto absoluto esperable para levetiracetam del 30%. El estudio fue positivo y el margen de no-inferioridad fue superado ampliamente, como era esperable, para el desenlace primario en la población “por protocolo”. La diferencia absoluta entre los dos grupos fue del 0,2%, bastante lejos del 15%. El aspecto más controvertido de este trabajo es el establecimiento arbitrario del margen de no-inferioridad, teniendo en cuenta que no se cumple, con la premisa de tener una muy buena medida de efecto en estudios previos (efecto de la CBZ-CR y del placebo) para esa población. Otra discusión es la duración del seguimiento y las dosis utilizadas. Algunos autores han denominado “diseños de pseudoplacebo” a los estudios de epilepsia en los que se utilizan dosis subóptimas como controles11.
Otro aspecto a considerar es el propio efecto placebo28,29. Así como puede variar la eficacia de un tratamiento de un estudio a otro por diferencias en el diseño, pacientes participantes, tratamientos concomitantes o simplemente por razones estadísticas, también puede variar el efecto placebo, y no necesariamente en la misma proporción que el tratamiento activo. Esto significa que la imputación al placebo se hace asumiendo que el efecto placebo es constante en el estudio actual de no-inferioridad respecto al/a los estudio/s de donde se toma el efecto del control activo.
Una potencial solución sería agregar una tercera rama placebo, pero esto no es posible en situaciones en las que ya hay una terapia estándar, particularmente en epilepsia o en prevención secundaria cerebrovascular. También es controvertida en Parkinson. Recientemente se llevó a cabo un estudio multicéntrico, multinacional de tres ramas entre un agonista dopaminérgico de uso corriente y eficacia demostrada contra una formulación de liberación extendida del mismo fármaco y otra rama placebo, todas sumado a selegilina en pacientes con Parkinson de novo, vírgenes de tratamiento con levodopa. En Argentina la Administración Nacional de Medicamentos y Tecnología Médica (ANMAT) obligó a modificar localmente el protocolo por considerar no ética la presencia de una rama placebo.
En Neurología el efecto placebo puede ser particularmente importante, por ejemplo en la enfermedad de Parkinson, en el dolor y en la depresión30,31.
En epilepsia, aunque un anticonvulsivante estándar tenga un efecto global establecido en la población, la magnitud del efecto comparada con placebo es baja en pacientes con alta y baja probabilidad de remisión de crisis, como por ejemplo pacientes con dos crisis en años separados y electroencefalograma (EEG) e imagen normal, y pacientes con 10 o más crisis, examen neurológico y EEG o imagen anormal respectivamente. Un estudio de no-inferioridad que reclute pacientes sesgados hacia uno u otro extremo del espectro puede encontrar la noinferioridad, pero debido a que ambos son ineficaces11.
No considerar el problema de la imputación al placebo puede llevar a otro fenómeno, conocido como “efecto arrastre” o “fenómeno de deriva” (bio-creep, phénoméne de dérive, “bioarrastre”): fenómeno por el cual la eficacia del producto investigado puede decaer a medida que se compara con controles cada vez menos eficaces. En la figura 4 vemos que si en un primer estudio se demuestra la superioridad de A sobre P, el tratamiento A puede transformarse en estándar y ser utilizado como control activo en un segundo estudio de no-inferioridad comparando B con A. Si se establece la no-inferioridad de B respecto de A y por alguna razón A deja de ser el tratamiento de referencia (por ejemplo evento adverso mortal idiosincrásico de baja frecuencia post-comercialización), entonces B podrá ser ahora el nuevo tratamiento disponible y por tanto el potencial control activo. En un tercer estudio C demuestra ser no-inferior a B, pero su real eficacia podría ser menor que la del placebo en el estudio original de superioridad8,9.

En la tabla 1 se resumen didácticamente las características metodológicas comparativas de los estudios de no-inferioridad y superioridad1.
Características metodológicas comparativas de los estudios de no-inferioridad y superioridad
| Objetivo del estudio | Superioridad | No inferioridad | |
| Tipo de control | Placebo | Activo | Control activo |
| Medir el tamaño del efecto absoluto contra placebo | Sí | No | No |
| Mostrar la existencia de un efecto | Sí | Sí | Probable |
| Comparar terapéuticas | No | Sí | Probable |
| Mostrar la sensibilidad del ensayo (assay sensitiuity) | Sí | Sí | No |
Las agencias regulatorias, incluyendo la European Medicines Agency (EMEA) o ANMAT, aceptan la evidencia proveniente de estudios de equivalencia y no-inferioridad para otorgar licencias para monoterapia32, mientras que la FDA no, particularmente en epilepsia. La FDA exige que para una monoterapia el nuevo tratamiento demuestre superioridad al menos sobre el placebo, lo que ha llevado a situaciones éticas de conflicto33. El estudio comentado de levetiracetam frente a carbamacepina fue el primero en cumplir con las regulaciones europeas vigentes para la evaluación de nuevos antiepilépticos para su indicación. En tales regulaciones se recomienda un estudio de no-inferioridad que muestre, al menos, similar relación riesgo-beneficio para el producto testado en comparación con el estándar conocido, usando dosis óptimas y desenlaces clínicamente relevantes. La guía estipula que el desenlace primario debería ser la proporción de pacientes libres de crisis por al menos 6 meses durante el período de evaluación, con un mantenimiento de la eficacia como mínimo de un año27,32.
Este visto bueno de la EMEA no se extiende a esclerosis múltiple donde “los estudios de equivalencia (no-inferioridad) son insuficientes como única prueba de eficacia, y se debería probar demostración de superioridad contra placebo o un comparador activo”. En una publicación (conference report) los autores escriben: “Tales estudios (de no-inferioridad) no proveen una evaluación de absoluta eficacia y seguridad, y no aportan una vía directa para establecer su sensibilidad (assay sensitivity). En ausencia de tal información, una conclusión de no-inferioridad no puede distinguir entre dos fármacos que son igual de eficaces o igual de ineficaces”34. No obstante, ha surgido también controversia al respecto35.
Hay un intenso debate alrededor de los ensayos de no-inferioridad36. Algunos autores afirman que no son éticos, independientemente de cualquier medida que se tome para sortear sus limitaciones metodológicas y de interpretación de resultados, y que la no necesidad de establecer superioridad cuando lo único que se desea es determinar una ventaja adicional, como una formulación de una toma diaria, es sólo un pretexto. Un punto de discusión en particular es la incapacidad de este tipo de diseño para detectar efectos adversos serios que sólo son evidentes en estudios más grandes, como los de superioridad y replicado en diferentes ensayos. Que “no hay límites al límite de no-inferioridad” y que por tanto, lo que es aceptable en términos estadísticos no lo es clínicamente. Por otra parte, si una razón para hacer estos estudios es disponer de tratamientos para grupos de pacientes que no responden al tratamiento convencional, entonces se está confundiendo un argumento válido con un abordaje incorrecto: ¿por qué no hacer estudios de superioridad para esos grupos de pacientes?37.
Desde un punto de vista comercial establecer la no-inferioridad de un nuevo producto es menos arriesgado que embarcarse en un estudio de superioridad en el que un resultado negativo dañaría la imagen del producto, aunque aportaría más información. Un estudio de no-inferioridad puede hacer pasar por alto información que haría que se detuviera la comercialización del producto. En definitiva, “deja al producto investigado en una especie de limbo: su lugar en la terapéutica no queda completamente establecido, pero su lugar en el mercado está asegurado”37.
En concordancia con esto, los estudios de no-inferioridad son interpretados como una argucia más en el camino de su introducción en el mercado, los llamados productos “yo también”, fármacos con pequeñas modificaciones respecto de la original, con alguna ventaja generalmente en su farmacocinética y/o preparación farmacéutica, permitiendo mantener porciones de mercado y extender patentes38.
Entre los argumentos a favor surge que en un mundo con cada vez menos recursos, los estudios de no-inferioridad pueden ayudar en determinadas patologías y zonas del planeta a acceder a tratamientos que de otra manera no estarían disponibles (particularmente para nuevos antibióticos en el tratamiento de tuberculosis), y que si bien es cierto que el establecimiento del margen de no-inferioridad es arbitrario, también lo es el error alfa del 5% utilizado en estudios de superioridad39.
ConclusiónDado que los estudios de no-inferioridad son cada vez más frecuentes en el ámbito de la Neurología y presentan aspectos metodológicos y éticos particulares, es necesario que los neurólogos tengamos conocimiento y participemos de la discusión para poder hacer una adecuada interpretación que nos permita estimar con claridad el real beneficio de una nueva terapia.
Conflicto de interesesEl autor ha participado como investigador principal en estudios de fase III para el laboratorio Boheringer Ingelheim y actualmente para Servier y Schering-Plough.
