




La hiperactividad simpática paroxística es una urgencia neurológica. Su fisiopatología aún no es clara y no existen estudios que respalden con evidencia óptima el tratamiento. La herramienta clínica desarrollada en el año 2014 ayuda a tener un grado de certeza diagnóstica y permite iniciar un manejo adecuado y oportuno. El desconocimiento de esta entidad conlleva al subdiagnóstico y al uso de intervenciones y exámenes innecesarios, aumentando la morbimortalidad en los pacientes. Presentamos dos casos clínicos, uno de la etiología más frecuente: trauma craneoencefálico, y el segundo, de una etiología muy poco común: síndrome de embolismo graso.
Paroxysmal sympathetic hyperactivity is a neurological emergency. Its pathophysiology is still unclear and there are no studies that support optimal treatment. The clinical tool developed in 2014 helps to have a degree of diagnostic certainty and allows the initiation of adequate and timely management. Ignorance of this entity leads to underdiagnosis and the use of unnecessary interventions and tests, increasing morbidity and mortality in patients. We present two clinical cases, one of the most frequent etiology: traumatic brain injury and the second, of a very rare etiology: fat embolism syndrome.
La hiperactividad simpática paroxística (HSP) se caracteriza por crisis recurrentes de taquipnea, taquicardia, diaforesis, hipertensión arterial (HTA), posturas motoras anómalas e hipertermia, que aparecen espontáneamente o en respuesta a estímulos externos, siempre de forma simultánea, en un paciente con una lesión cerebral previa1,2.
En el año 2014, un consenso de expertos definió los criterios diagnósticos y decidió utilizar el término HSP como el concepto que unifica al menos a 31 denominaciones usadas hasta entonces para esta patología2.
Aproximadamente de 7 a 33% de los pacientes ingresados a una Unidad de Cuidados Intensivos (UCI) posterior a una lesión cerebral grave pueden desarrollar HSP3. Las etiologías más frecuentes son el trauma craneoencefálico (TCE), la encefalopatía hipóxico-isquémica y la enfermedad cerebrovascular (ECV). Dentro de otras entidades, el síndrome de embolismo graso (SEG) es una causa muy poco común4,5.
A continuación, presentamos dos casos y una breve revisión de la literatura.
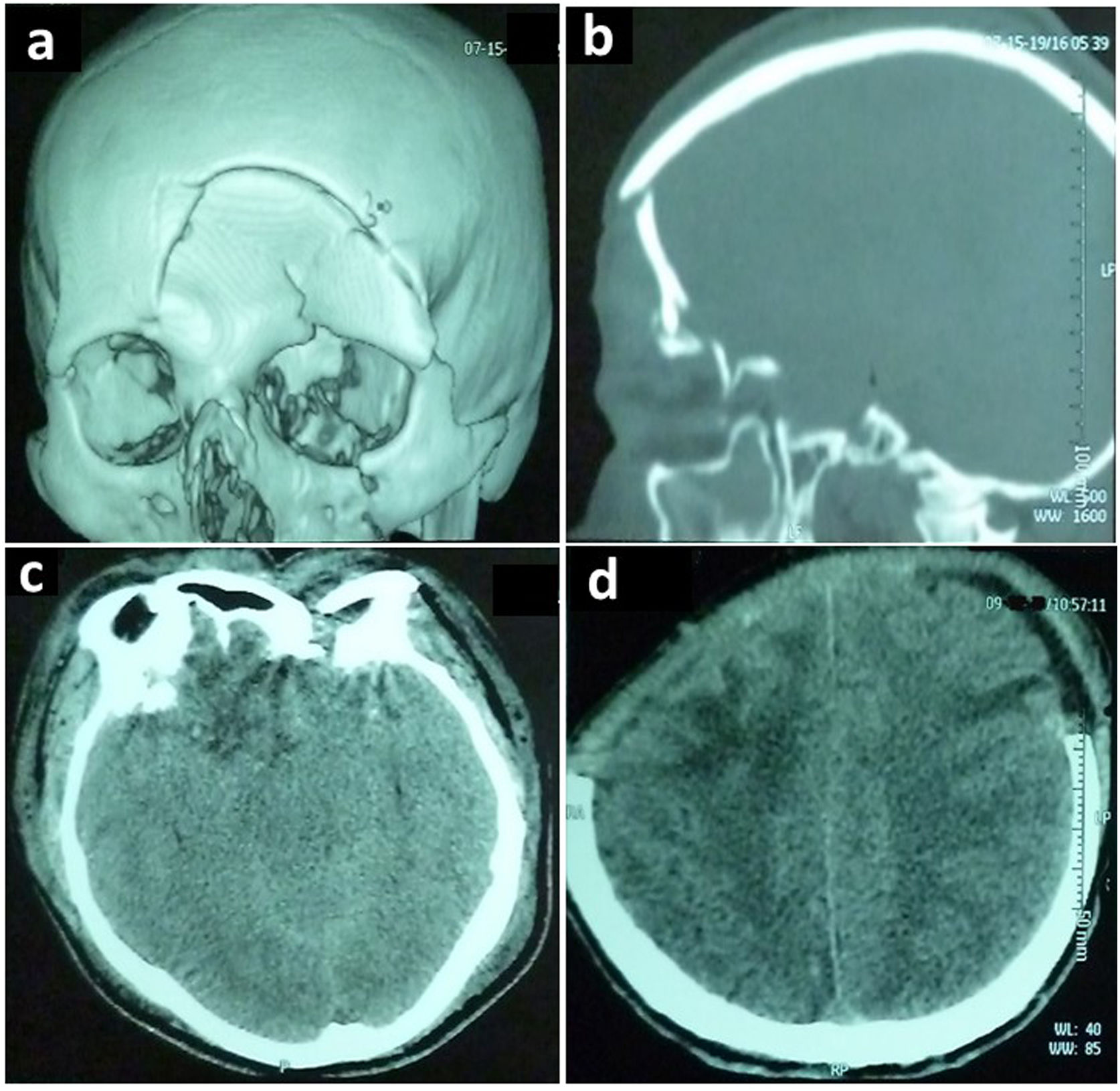
Casos clínicosCaso clínico 1Paciente masculino de 34 años, sin antecedentes, ingresó posterior a sufrir un accidente de tránsito. La exploración física evidenció anisocoria, escala de coma de Glasgow (ECG) 6/15 y fractura del hueso frontal (fig. 1). Requirió una craneotomía para evacuación de los hematomas, cierre de una fistula de líquido cefalorraquídeo (LCR) traumática y colocación de un captor de presión intracraneal (PIC). Ingresó a UCI bajo neuroprotección y cobertura antibiótica.
 en ventana ósea una fractura frontal hundida, fractura del techo y piso de la órbita izquierda. En ventana parenquimatosa: (c) edema cerebral difuso con borramiento de las cisternas de la base, contusiones hemorrágicas a nivel orbito-frontal derecho e izquierdo. (d) Craneotomía bilateral anterior.")
Tomografía computarizada de cerebro sin contraste, correspondiente al primer caso descrito, que demuestra: (a, b) en ventana ósea una fractura frontal hundida, fractura del techo y piso de la órbita izquierda. En ventana parenquimatosa: (c) edema cerebral difuso con borramiento de las cisternas de la base, contusiones hemorrágicas a nivel orbito-frontal derecho e izquierdo. (d) Craneotomía bilateral anterior.
Con valores de PIC normales se retiró el captor y se suspendió la sedación logrando una ECG 10/15.
A los 28 días de evolución se reportaron cifras de tensión arterial fluctuantes (hasta 150/100), diaforesis, taquicardia (110 lpm), taquipnea (23 rpm) y fiebre (39°), duraban aproximadamente 5 min, y en ocasiones se desencadenaban por cambios de posición. Se consideró una respuesta inflamatoria sistémica secundaria a una colección parietotemporal derecha. Fue manejado con limpiezas quirúrgicas y antimicrobianos por 14 días (vancomicina y fluconazol).
Las crisis disautonómicas no cedieron y se sumó rigidez muscular generalizada llegando a postura de opistótonos. Los cultivos fueron negativos (incluyendo LCR). El electroencefalograma demostró actividad lenta difusa en relación con un proceso de afectación encefálica moderada, con actividad lesional bifrontal, descartándose actividad epileptiforme. Se planteó el diagnóstico de HSP, se inició propranolol y gabapentina con respuesta parcial. La adición de clonazepam logró un mejor control y se mantuvo sin crisis de disautonomía los días posteriores. Fue derivado a una unidad de cuidado crónico, donde finalmente falleció un mes posterior a su alta.
Caso clínico 2Paciente masculino de 22 años, previamente sano, víctima de un accidente de tránsito. El examen físico evidenció ECG 15/15 y fracturas de fémur, tibia y peroné derechos, que fueron resueltas quirúrgicamente.
Cinco horas posteriores al ingreso, se observó un deterioro neurológico súbito con ECG 9/15. Ingresó a UCI bajo sedación. Se evidenciaron petequias subconjuntivales y en el tórax anterior, hombros y extremidades. La tomografía cerebral inicial no demostró lesiones agudas.
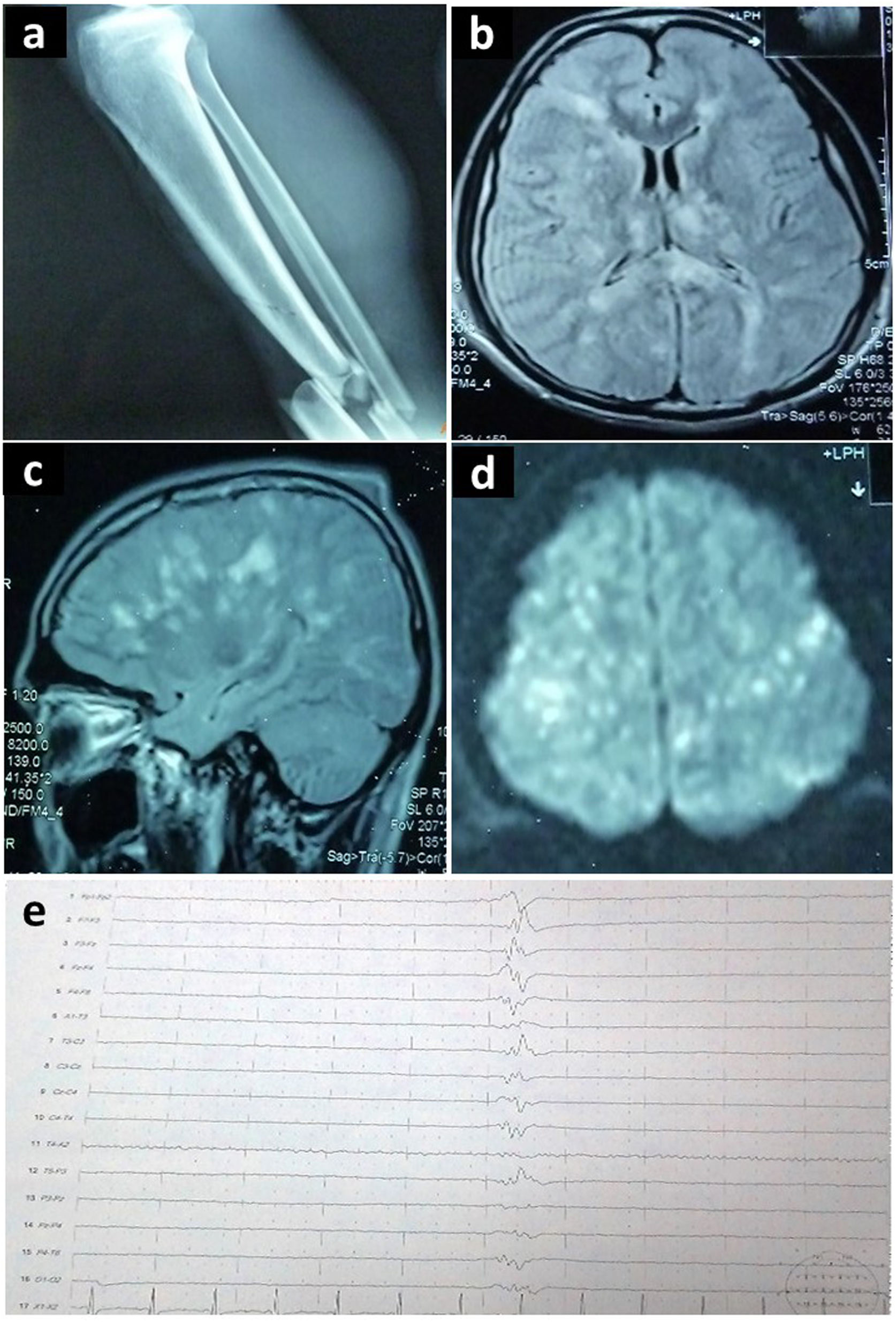
A las 72 horas presentó una crisis convulsiva tónico-clónica generalizada, por lo que se optimizó la sedación y se inició fenitoína. Se realizaron estudios complementarios (fig. 2).
 Radiografía de miembro inferior derecho que demuestra fractura de tibia y peroné. (b, c, d) Resonancia magnética cerebral en corte axial y sagital, que demuestra en secuencia FLAIR focos hiperintensos en sustancia blanca, tálamos y cuerpo calloso, afectando múltiples territorios vasculares (patrón de campo de estrellas). (e) Electroencefalograma que demuestra patrón de brote-supresión.")
Estudios complementarios realizados en el segundo caso descrito: (a) Radiografía de miembro inferior derecho que demuestra fractura de tibia y peroné. (b, c, d) Resonancia magnética cerebral en corte axial y sagital, que demuestra en secuencia FLAIR focos hiperintensos en sustancia blanca, tálamos y cuerpo calloso, afectando múltiples territorios vasculares (patrón de campo de estrellas). (e) Electroencefalograma que demuestra patrón de brote-supresión.
El ecocardiograma evidenció un foramen oval permeable y se diagnosticó SEG. Las crisis convulsivas persistieron pese a los fármacos antiepilépticos (levetiracetam y lamotrigina). Se empleó tiopental, logrando un patrón de brote-supresión en el electroencefalograma (fig. 2e). Al día 17 se retiró el barbitúrico. Hubo apertura ocular espontánea, debilidad generalizada y crisis recurrentes de HTA (160/110), taquicardia (120 lpm), taquipnea (22 rpm), diaforesis y temblor en miembros inferiores. Se presentaban hasta tres episodios diarios y duraban en promedio 10 minutos. El electroencefalograma no demostró actividad epileptiforme. Los cultivos fueron negativos.
Se consideró HSP, se inició propranolol y morfina. Las crisis persistieron y se desencadenaban por la aspiración de secreciones. Se decidió adicionar gabapentina, con mejor respuesta. Al día 45 del evento, sin evidencia de crisis o infecciones, fue transferido a un centro de cuidado crónico, donde falleció a los 60 días de su externación.
DiscusiónEstudios retrospectivos han identificado las principales patologías predisponentes al desarrollo de HSP. Las lesiones cerebrales traumáticas se asocian con mayor frecuencia, en comparación con lesiones cerebrales no traumáticas, con una relación aproximada de 4-5:16.
El TCE grave es el principal desencadenante de HSP (80%), le siguen la encefalopatía hipóxico-isquémica (9,7%) y la ECV (4-5%). En cuanto a ECV, la hemorragia intraparenquimatosa (> 60 mL) prevalece sobre la hemorragia subaracnoidea y el ictus isquémico. La hidrocefalia, la encefalitis autoinmune, los tumores e infecciones del sistema nervioso central (SNC), en conjunto representan < 5% de los casos1,6,7.
El SEG es una complicación grave que se presenta con mayor frecuencia en pacientes con fracturas cerradas y multifragmentarias de los huesos largos de las extremidades inferiores, aunque se describen otras etiologías8. Su incidencia varía de 1 a 15%, según la causa9,10. Clínicamente se caracteriza por la tríada de lesión pulmonar aguda, hemorragia petequial y alteraciones neurológicas11,12. La HSP secundaria a SEG es rara, se conocen cuatro reportes de esta asociación5,12–14.
Varias teorías intentan explicar la respuesta disautonómica característica de la HSP. Modelos animales y estudios de neuroimagen funcional han permitido descartar algunas de ellas e identificar las estructuras probablemente involucradas, sin lograr dilucidar por completo la fisiopatología4,7.
El sistema nervioso (SN) autónomo es el encargado del control homeostático del organismo. La respuesta ante estímulos internos y medioambientales provenientes de la médula espinal (ME) es regulada por centros superiores en el SNC. Fisiológicamente ante una lesión, la activación del SN simpático y la consecuente secreción de catecolaminas (respuesta excitatoria), intentan revertir los efectos adversos de la injuria. En segundos, se duplica la frecuencia cardiaca y la presión arterial, asegurando un flujo sanguíneo adecuado hacia los órganos vitales para evitar un daño secundario3,6. La inhibición del sistema simpático por los centros superiores y la activación del parasimpático restauran la homeostasis. La pérdida de esta regulación deriva en una actividad simpática anómala15.
La teoría de la relación excitación:inhibición considera al diencéfalo y al tronco cerebral (especialmente la sustancia gris periacueductal), como los centros superiores inhibidores. La lesión en estos centros o sus conexiones conlleva a la estimulación de los circuitos medulares, la sensibilización en el asta posterior de la ME y la amplificación de los estímulos, desarrollando alodinia1,6. Esto explica cómo los estímulos ambientales (succión de secreciones, baño, cambios de posición) o aferencias viscerales (retención urinaria, constipación) provocan la activación del SN simpático y desencadenan los signos cardinales de la HSP3,16.
El diagnóstico es clínico, requiere una alta sospecha y el descarte de otras patologías con características similares (sepsis, crisis convulsivas, síndrome neuroléptico maligno, hidrocefalia)3–5.
Se han reconocido tres etapas clínicas del síndrome:
Primera etapa: dura aproximadamente siete días, generalmente sin signos de disautonomía porque los pacientes permanecen bajo sedación profunda, como parte de su manejo16,17.
Segunda etapa: inicia desde la segunda semana de evolución y se extiende en promedio hasta los 74 días. Los signos clínicos se expresan ampliamente. Aquí se identifican la mayor parte de casos. El cese de los episodios de diaforesis marca el fin de esta etapa17.
Tercera etapa: puede prolongarse por varios meses y las crisis son menos frecuentes. Puede evidenciarse distonía o espasticidad en muchos pacientes, pero no está claro si son complicaciones del síndrome o consecuencia de las lesiones cerebrales1,17,18.
En los dos casos se reconocieron las dos primeras etapas. En el segundo paciente, las crisis aparecieron después del retiro de la sedación profunda y se repitieron en los días siguientes hasta su diagnóstico.
La PSH-AM es un instrumento que permite tener un grado de certeza diagnóstica. Consta de dos partes: la escala de características clínicas (CFS), que valora la severidad de los signos cardinales de la HSP (taquipnea, taquicardia, diaforesis, hipertensión arterial, posturas motoras anómalas e hipertermia), otorgándoles un valor de 0 (ausente) a 3 puntos (severo) a cada uno; y la herramienta de probabilidad diagnóstica (DLT), que evalúa la probabilidad de HSP en los pacientes, de acuerdo con los rasgos de presentación del cuadro (un punto por cada característica presente)2. La suma de las dos otorga la probabilidad de diagnóstico de HSP en: improbable < 8 puntos, posible 8-16 puntos y probable > 17 puntos1,2. En el primer caso, la CFS puntuó 14 (severo) y en el segundo 9 (moderado). El puntaje total de PSH-AM fue 25 en el primero y 17 en el segundo paciente; es decir, HSP probable en ambos casos.
Los objetivos del tratamiento son: bloquear el estímulo simpático central, disminuir la alodinia y reducir el efecto periférico de las catecolaminas (evitar el daño terciario). No existe un protocolo de manejo estandarizado. La mayoría de las recomendaciones se basan en series de casos publicados, sin nivel de evidencia óptimo1,16. Se acepta que el abordaje apropiado implica medidas farmacológicas y no farmacológicas3.
Las recomendaciones no farmacológicas incluyen:
- -
Minimizar los estímulos externos
- -
Intervención nutricional adecuada (la estimulación simpática produce un mayor gasto energético que puede llevar a la pérdida de peso y desnutrición).
- -
Fisioterapia temprana con hincapié en la movilidad activa y pasiva buscando evitar la rigidez y contracturas musculares.
En los dos casos, las crisis se desencadenaban por estímulos externos (aspiración de secreciones y cambios de posición), por lo que se minimizaron los mismos.
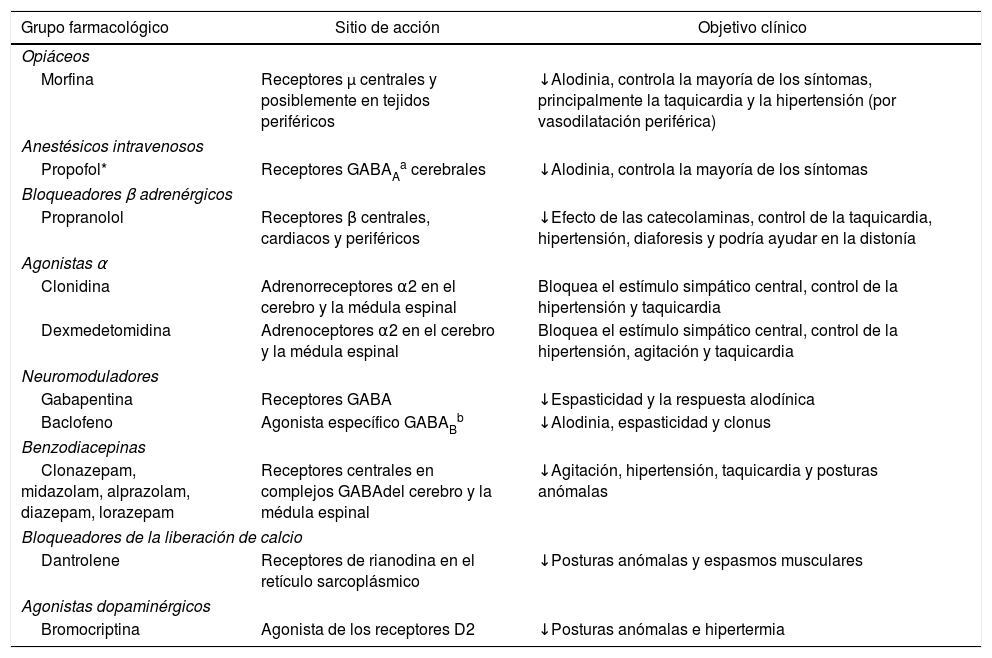
La asociación de al menos dos grupos farmacológicos es superior a la monoterapia para el control de las crisis1,4,18. Los principales fármacos descritos en el tratamiento de HSP se resumen en la tabla 1.
Principales fármacos utilizados para el tratamiento de la hiperactividad simpática paroxística
| Grupo farmacológico | Sitio de acción | Objetivo clínico |
|---|---|---|
| Opiáceos | ||
| Morfina | Receptores μ centrales y posiblemente en tejidos periféricos | ↓Alodinia, controla la mayoría de los síntomas, principalmente la taquicardia y la hipertensión (por vasodilatación periférica) |
| Anestésicos intravenosos | ||
| Propofol* | Receptores GABAAa cerebrales | ↓Alodinia, controla la mayoría de los síntomas |
| Bloqueadores β adrenérgicos | ||
| Propranolol | Receptores β centrales, cardiacos y periféricos | ↓Efecto de las catecolaminas, control de la taquicardia, hipertensión, diaforesis y podría ayudar en la distonía |
| Agonistas α | ||
| Clonidina | Adrenorreceptores α2 en el cerebro y la médula espinal | Bloquea el estímulo simpático central, control de la hipertensión y taquicardia |
| Dexmedetomidina | Adrenoceptores α2 en el cerebro y la médula espinal | Bloquea el estímulo simpático central, control de la hipertensión, agitación y taquicardia |
| Neuromoduladores | ||
| Gabapentina | Receptores GABA | ↓Espasticidad y la respuesta alodínica |
| Baclofeno | Agonista específico GABABb | ↓Alodinia, espasticidad y clonus |
| Benzodiacepinas | ||
| Clonazepam, midazolam, alprazolam, diazepam, lorazepam | Receptores centrales en complejos GABAdel cerebro y la médula espinal | ↓Agitación, hipertensión, taquicardia y posturas anómalas |
| Bloqueadores de la liberación de calcio | ||
| Dantrolene | Receptores de rianodina en el retículo sarcoplásmico | ↓Posturas anómalas y espasmos musculares |
| Agonistas dopaminérgicos | ||
| Bromocriptina | Agonista de los receptores D2 | ↓Posturas anómalas e hipertermia |
Los dos pacientes recibieron propranolol como primera opción, pero la asociación de otros grupos farmacológicos logró una mejor respuesta.
ConclusionesLa HSP es una condición potencialmente letal y su fisiopatología aun no es clara. La detección temprana evita intervenciones inadecuadas y disminuye las complicaciones. El manejo multidisciplinario, las medidas no farmacológicas y el uso de al menos dos grupos farmacológicos para el control de las crisis se asocian con una mejor respuesta.
Consideraciones éticasEl protocolo para la realización del presente trabajo fue presentado y aprobado por el Comité de Ética de Investigación en Seres Humanos (CEISH) del Hospital de Especialidades Carlos Andrade Marín, autorizando el uso de la información. Se garantizó el anonimato total de los pacientes así como la protección de sus datos personales.
FinanciaciónLa presente investigación no ha recibido ayudas específicas provenientes de agencias del sector público, sector comercial o entidades sin ánimo de lucro.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
