




La enfermedad de Fabry es una enfermedad por depósito lisosomal, con herencia ligada al cromosoma X, causada por la deficiencia de la enzima α-galactosidasa A, lo que resulta en la acumulación de glucoesfingolípidos en distintos tipos celulares. La terapia de reemplazo enzimático específica ha demostrado revertir algunos síntomas y disminuir la chance de eventos renales, cardíacos y cerebrovasculares.
ObjetivoPresentar la evolución de un paciente con enfermedad de Fabry tras la interrupción de la terapia de reemplazo enzimático.
Caso clínicoPaciente masculino con diagnóstico confirmado de enfermedad de Fabry por métodos bioquímicos y test genético, que inicia tratamiento con agalsidasa beta, presentando dolor neuropático en cuatro miembros, compromiso disautonómico, hipohidrosis e insuficiencia renal, con proteinuria nefrótica, sin compromiso cardíaco y ausencia de signos neurológicos centrales. La resonancia magnética cerebral mostraba una isquemia lacunar periventricular derecha. Tras 2 años de respuesta favorable a la terapia de reemplazo enzimático, el paciente suspende el tratamiento. Se presenta dos años después con aumento del dolor neuropático, compromiso cardíaco y nuevas imágenes isquémicas en la sustancia blanca.
ConclusiónEl inicio del tratamiento con terapia de reemplazo enzimático, aun en etapas avanzadas de la enfermedad, mostró una respuesta favorable, y la interrupción del tratamiento es deletérea, mostrando un rápido reinicio de los síntomas propios de la enfermedad de Fabry.
Fabry disease is a X-linked lysosomal storage disease caused by an inborn deficiency of α-galactosidase A, which results in progressive accumulation of globotriaosylceramide and other neutral glycolipids in different cells and tissues. Enzyme replacement therapy showed symptoms reversion and decreased the possibility of renal, cardiac and cerebrovascular events.
AimTo present the follow-up of Fabry patient after enzyme replacement therapy interruption.
Case reportMale patient with confirmed diagnosis of Fabry disease by chemical and genetic testing, who started treatment with agalsidasa Beta for neuropathic pain in four limbs, autonomic involvement, hypohidrosis and renal failure, without cardiac compromise and lack of central nervous system signs. Magnetic resonance image did show one lacunar stroke in paraventricular right region. After two years of good response to enzyme replacement therapy, patient decided to suspend treatment. He presented two years later for increasing pain, cardiac involvement and new ischemic strokes in white matter.
ConclusionEnzyme replacement therapy can improve symptoms even in advanced stages of the disease and the treatment suspension shows a fast deterioration of impaired organs and the new appearance of Fabry signs.
La enfermedad de Fabry (EF) es una enfermedad por depósito lisosomal, con herencia ligada al cromosoma X, causada por la deficiencia de la enzima α-galactosidasa A (A-GalA)1; la falta de esta enzima resulta en la acumulación de glucoesfingolípidos, preferentemente globotriaosilceramida (Gl3) en células endoteliales, periteliales, musculares lisas de los vasos sanguíneos y estructuras del sistema nervioso central y periférico2. La EF está distribuida en todo el mundo, con una incidencia que varía entre 1/40.0001 y 1/117.0003. Desde el año 2001 se dispone de una terapia de reemplazo enzimático (TRE) específica, que ha demostrado revertir algunos síntomas y disminuir la chance de eventos renales, cardíacos y cerebrovasculares, todo dependiendo del momento de su inicio4,5. Los primeros síntomas se expresan durante la niñez con dolor distal de tipo neuropático en los cuatro miembros e hipohidrosis, asociado a lesiones cutáneas conocidas como angioqueratomas. Durante la adolescencia se agrega depósito de Gl3 en la córnea (córnea verticilada), manifestaciones disautonómicas, fatiga y disminución de la capacidad auditiva, y llegado el estado adulto, se desarrolla insuficiencia renal y cardíaca, y aparecen también accidentes cerebrovas- culares de repetición. El compromiso renal en hombres (hemicigotos) y mujeres (heterocigotas) se desarrolla desde la segunda y tercera década, respectivamente, existiendo gran variabilidad en su presentación6. Los hallazgos histológicos en la biopsia renal muestran el depósito de Gl3 en podocitos, células mesangiales, tubulares (preferentemente en túbulos distales) y vasculares (endotelio arterial, venoso, capilar y en células musculares lisas de la pared)7.
ObjetivoPresentar la evolución multisistémica de un paciente con EF tras la interrupción de la TRE.
Caso clínicoPaciente de sexo masculino que a los 8 años de edad comienza con dolor de características neuropáticas a nivel distal en los cuatro miembros, con aumento de su intensidad ante la fiebre, los cambios de temperatura ambiental y el ejercicio. Asociado al dolor, se describe menor sudoración en comparación con otros niños (hipohidrosis). Durante la adolescencia presenta lesiones dérmicas puntiformes milimétricas (1 a 2mm) a nivel genital y proximal de miembros inferiores (angioqueratomas), asociados a astenia, fatiga muscular y trastornos digestivos, caracterizados por diarreas y dolores abdominales de tipo cólico. A los 22 años, ante la presencia de insuficiencia renal, se sospecha EF, por lo que se realiza dosaje de A-GalA con resultado 0,8nmol/h/mg (normal: 30,5-57,7nmol/h/mg) y test genético, que informa mutación C223A.
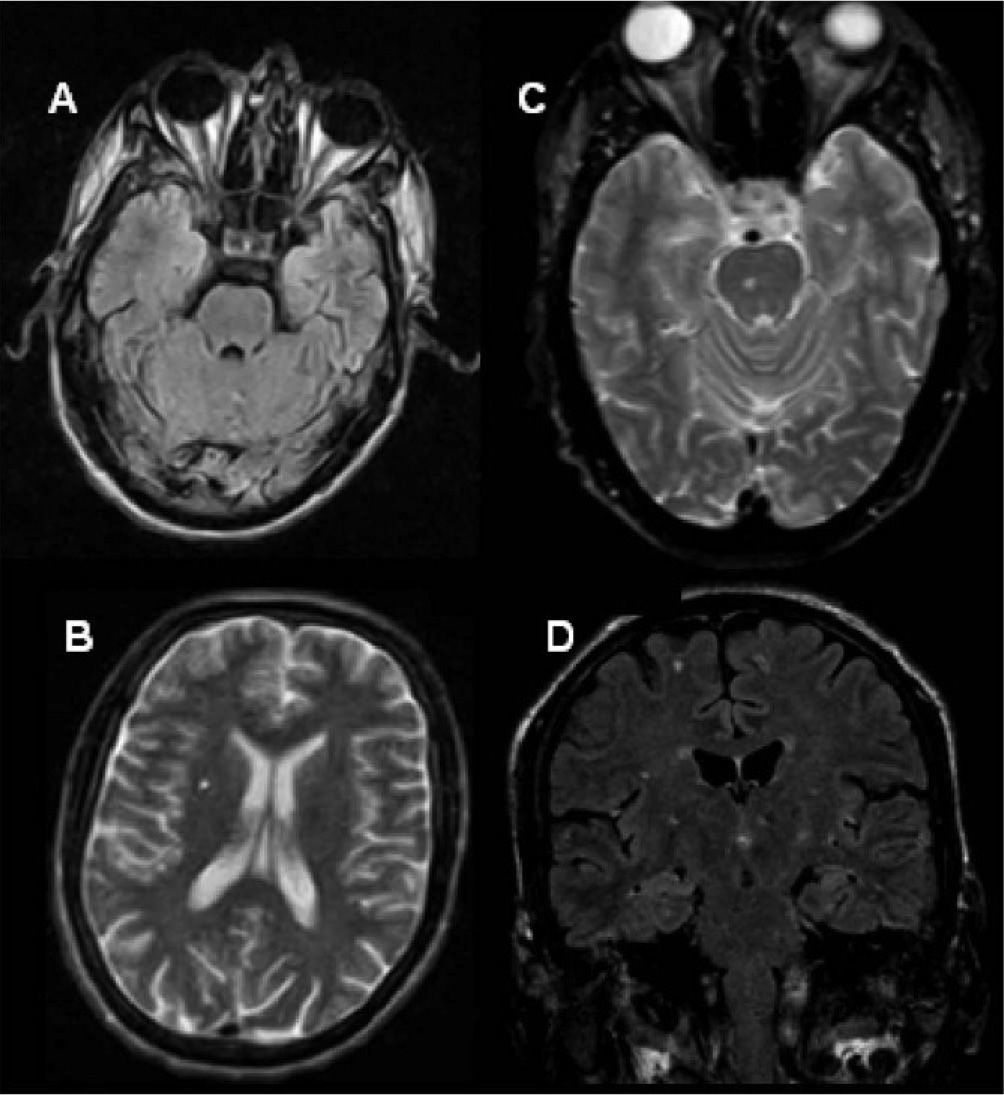
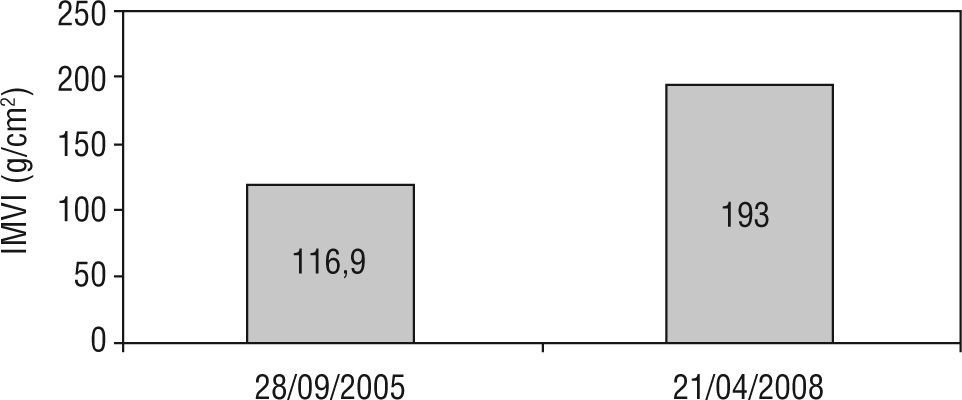
A los 27 años de edad (septiembre de 2002) se inicia TRE con agalsidasa beta en dosis de 1mg/kg cada 2 semanas. La evaluación nefrológica al inicio del tratamiento arrojó: insuficiencia renal en estadio 3 con proteinuria en rango nefrótico. La resonancia magnética cerebral mostró una única lesión lacunar a nivel periventricular derecho (fig. 1). Desde el punto de vista cardiológico el electrocardiograma no mostró alteraciones y el ecocardiograma bidimensional informó grosor parietal posterior del ventrículo izquierdo y septum interventricular normales (10 y 11mm, respectivamente) (fig. 2) e índice de masa ventricular izquierda (IMVI) de 116,9g/m2(valor normal, <131g/m2). Las válvulas mitral y aórtica presentaban ligero engrosamiento sin alteraciones hemodinámicas. Las carótidas mostraban grosor miointimal normal y ausencia de obstrucciones. El estudio de vasodilatación mediada por flujo realizado en la arteria humeral izquierda con eco-doppler mostraba una respuesta nula a la hiperemia, con ausencia de vasodilatación, lo que representaba disfunción endotelial.
 Lesión isquémica única lenticular derecha antes del inicio de la terapia de reemplazo enzimático (TRE). C-D) Múltiples imágenes isquémicas a nivel supra e infratentorial no presentes en la imagen anterior.")
 tras la interrupción de la terapia de reemplazo enzimático (TRE).")
Tras 2 años de TRE sin interrupciones, el paciente refirió mejoría de los síntomas digestivos, incremento de la sudoración, desaparición de la fatiga muscular y mejoría del dolor neuropático. La insuficiencia renal progresó a estadio 4, requiriendo hemodiálisis a partir del 2005. Los estudios cardiológicos y la evaluación neurológica mostraban estabilidad, sin signos de compromiso progresivo. El paciente decidió suspender los controles y la TRE, continuando la hemodiálisis en forma adecuada.
Dos años después el paciente se presenta a control con reinicio del dolor neuropático, requiriendo tratamiento con morfina (jarabe) en forma intermitente, fatiga muscular severa y un evento de hemiparesia braquiocrural derecha súbito autolimitado diez días previo a la nueva consulta. Un nuevo ecocardiograma mostró incremento del grosor parietal de la pared posterior del ventrículo izquierdo y del septum interventricular (13 y 15mm, respectivamente) y el IMVI se incrementó un 60,6% (de 116,9 a 193g/m2) (fig. 2). Una nueva resonancia magnética cerebral mostró múltiples lesiones isquémicas (fig. 1). El paciente decide iniciar nuevamente la TRE con agalsidasa beta.
ComentarioLa EF es una enfermedad progresiva que afecta varios órganos y sistemas y donde la TRE ha demostrado cambiar el curso natural de la enfermedad. Los primeros estudios que llevaron a la aprobación de la agalsidasa beta para el tratamiento de la EF en el año 2001 se realizaron con pacientes con escasa afección renal y fueron excluidos los que presentaban creatinina sérica >2,2mg/dl, necesidad de diálisis o trasplante renal. Esta población mostró que, tras 5 meses de tratamiento con dosis de 1mg/kg, se producía una remoción completa del Gl3 de las células del endotelio capilar renal, medido en la biopsia renal previa y posterior al tratamiento4. Estos resultados fueron reproducidos y mantenidos tras 54 meses de tratamiento8. Debido a que no se disponía de resultados en pacientes con afección marcada a nivel cardíaco, renal y cerebrovascular, se realizó un estudio que incluía a este grupo de pacientes con EF avanzada6. El resultado mostró que el inicio de TRE retrasaba la aparición de eventos renales, cardíacos y cerebrovasculares, en comparación con el grupo placebo. En estos últimos dos reportes se definieron algunos marcadores de mal pronóstico renal, como la presencia de proteinuria >1g/día, aclaramiento de creatinina ≤55ml/min × 1,73m2, presencia de esclerosis glomerular >50% de la muestra y creatinina sérica >1,5mg/dl.
El paciente presentado mostró una respuesta positiva al dolor neuropático, a la fatiga, a las manifestaciones gastrointestinales y a la hipohidrosis. Durante los dos años de TRE, la evaluación cardiológica así como los estudios ecocardiográficos no mostraron progresión de la EF. La función renal presentó una mala evolución, requiriendo iniciar hemodiálisis. Esta situación fue congruente con los estudios previamente citados.
No existen a la fecha reportes en los que se presente la evolución de los pacientes con EF tras la interrupción de la TRE. Este caso nos permite concluir que aun ante el inicio tardío del tratamiento con TRE, el paciente mostró buena evolución general, y que la interrupción del tratamiento es deletérea, mostrando un rápido reinicio de los síntomas propios de la EF.
Conflicto de interesesLos autores declaran que no tienen ningún conflicto de intereses.
