




Las enfermedades mitocondriales incluyen un amplio espectro de patologías muchas de ellas causadas por el compromiso de la cadena respiratoria mitocondrial y por ende del metabolismo energético.
ObjetivoEl propósito de este artículo es realizar una revisión de los cuadros clínicos causados por mutaciones en los genes implicados en la regulación y la reparación de ADN mitocondrial (ADNmt), con especial énfasis en los genes POLG1 y 2.
DesarrolloHasta no hace mucho tiempo, mutaciones en genes nucleares que afectaban indirectamente el ADNmt estaban vinculadas casi exclusivamente a cuadros severos de inicio infantil. Sin embargo, tras la descripción de casos en adultos con formas de herencia mendeliana o incluso esporádica por mutaciones en genes nucleares implicados en la reparación y control del ADNmt, se ha abierto un nuevo capítulo en la llamada “medicina mitocondrial”. Las más destacadas son las producidas en POLG1 dentro de los trastornos debidos a alteración en la estabilidad mitocondrial.
ConclusionesLas mutaciones en el gen POLG1 que codifica para la enzima mitocondrial POLG γ se han convertido en una causa frecuente de mutaciones tanto en niños como en pacientes adultos, dando lugar al reconocimiento de fenotipos relativamente específicos.
Mitochondrial diseases include a wide spectrum of disorders, most of them due to defects of the mitochondrial respiratory chain and therefore impairment of energetic metabolism.
ObjectiveThe aim of this review is to make an update of the clinical entities related with mutations in genes involved in regulation and repair of the mitochondrial DNA (mtDNA), especially POLG1 and POLG2 gene mutations.
DiscussionNot long time ago, mutations in nuclear genes which affect indirectly mtDNA, were exclusively associated with severe infantile-onset diseases. However, since the description of autosomal dominant or even sporadic mutations in adult cases, a new chapter in mitochondrial medicine has been developed. POLG1 mutations became the most frequent and relevant gene in the so called “disorders due to gene defects altering the stability of mtDNA”.
ConclusionsPOLG1, which encodes for the POLG γ mitochondrial enzyme, has become a frequent cause of mitochondrial diseases, not only in children but also in adults, given birth to the recognition of specific phenotypes.
La cadena respiratoria yace en la membrana interna de la mitocondria y tiene por finalidad la producción de energía a través de la síntesis de adenosín trifosfato (ATP)1–3. De los 93 genes conocidos que participan en la síntesis de los elementos constitutivos de la misma, sólo 13 están sintetizados por el ADN mitocondrial (ADNmt), siendo el resto dependiente del ADN nuclear (ADNn). A su vez, todos los componentes de las enzimas que conforman el sistema de reparación y síntesis del ADNmt (helicasa o “Twinkle”, POLG γ, adenosín translocasa o ANT1, entre otras) son sintetizados por genes nucleares4,5. El ADNmt está formado por una cadena circular de 16.569 pares de bases, sin histonas ni intrones, y a diferencia del ADN nuclear se encuentra en constante replicación, siendo por estos motivos plausible de presentar frecuentes mutaciones6. Una de las enzimas responsables es la polimerasa-γ, y su mal funcionamiento es causa de numerosos cuadros clínicos que se describen en la presente revisión.
Aspectos genéticos y molecularesPOLG γ y el gen POLGLa holoenzima humana POLG γ es un heterotrímero compuesto por una subunidad principal (subunidad catalítica) y dos subunidades accesorias idénticas7. Es la única polimerasa de ADN conocida ubicada dentro de la mitocondria, y junto a la proteína mitocondrial de unión de cadena simple (mtSSB, single-stranded binding protein) y la helicasa “Twinkle” conforman el replisoma humano mitocondrial8,9. El precursor proteico de POLG γ es sintetizado en el citosol, y tras su clivaje es importado a la matriz mitocondrial. La proteína madura tiene un peso molecular de 140kDa (p140) y está dividida en tres dominios o regiones: a) la región 3’-5’ exonucleasa; b) la región de unión, y c) la región carboxiterminal altamente conservada o polimerasa10,11. En las tres regiones se han descrito mutaciones y variantes polimórficas, las cuales son codificadas por el gen POLG1. Las dos subunidades accesorias de POLG γ poseen un peso molecular de 55kDa (p55) y participan incrementando la afinidad de unión de la subunidad principal al ADNmt durante la replicación12 y son codificadas por el gen POLG213.
El gen humano POLG o POLG1 fue identificado en el año 1996 y su primera mutación cinco años después, en un paciente con oftalmoplejía crónica progresiva (CPEO, chronic progressive external ophthalmoplegia) y múltiples deleciones en el ADNmt14. Se encuentra ubicado en el cromosoma 15q25 y está compuesto por 23 exones con el inicio de codón codificante (Met1) en el exón 2. El exón 2, a su vez, posee una secuencia repetitiva de 10 tripletes CAG que codifican para un tracto poliglutamínico (poliQ). Se han descrito variaciones en el número de repeticiones CAG, asociadas a ciertas condiciones como la enfermedad de Parkinson15,16, el cáncer testicular17 y la infertilidad masculina18, aunque se necesitan más estudios para establecer una relación causal entre el número de repeticiones de poliQ y las tres condiciones previamente mencionadas. Prueba de ello han sido ciertos estudios in vitro, los cuales no han podido reproducir los resultados obtenidos, poniendo en duda el rol patogénico de POLG1 en estas enfermedades19,20.
Por otra parte, en el año 2006 se describió la primera mutación en POLG221, gen que codifica para las dos subunidades accesorias, ubicado en el cromosoma 17q21.
Mutaciones y polimorfismos en POLG1 y POLG2Desde la primera mutación descrita en POLG1, más de 150 entidades clínicas han sido vinculadas a mutaciones en ambos genes con una distribución uniforme en las tres regiones del gen POLG1. Sin embargo, no se ha podido encontrar una estrecha correlación geno-fenotípica22 entre el tipo y la ubicación de la mutación y los síndromes clínicos ni la razón de por qué la misma mutación puede generar fenotipos completamente distintos. A pesar de ello, la mayoría de las mutaciones que se presentan con un patrón autosómico dominante (p. ej., p.Y955C en adCPEO; véase más adelante) se ubican en la región polimerasa, mientras que las formas recesivas habitualmente lo hacen en heterocigosis, con una mutación en la región de unión y otra en la región polimerasa (p. ej., síndrome de Alpers). La mutación p.A467T (cambio de una alanina a treonina en la posición 467 de la secuencia proteica) es la mutación más frecuentemente hallada en los tres principales síndromes clínicos POLG-asociados (CPEO, síndromes ataxia-neuropatía y síndrome de Alpers). Habitualmente esta mutación está vinculada a formas recesivas, aunque se han descrito formas CPEO de inicio tardío de tipo autosómico dominante23.
Existen ciertos polimorfismos que no ejercerían cambios funcionales en la proteína (p. ej., p.P18S o p.P241L). Sin embargo, hay otro grupo de polimorfismos que podrían ejercer un rol modulador frente a la presencia de otras mutaciones, disminuyendo la actividad catalítica de la enzima. Tal es el caso de c.3428A>G p.E1143G, un polimorfismo frecuentemente hallado en la población caucásica pero exageradamente asociado con otras mutaciones24. No se han descrito patologías vinculadas a cambios en homocigosis de E1143G, reforzando la idea de su falta de patogenicidad per se. Sin embargo, se han hallado numerosas condiciones donde la misma se encuentra en trans o cis junto a otras mutaciones25. Es decir, podría modificar la expresión de otras mutaciones.
Finalmente se han descrito mutaciones en POLG γ (p.R579W+p.A889T) que, de forma excepcional, pueden asociarse a formas esporádicas de CPEO26.
Actualmente existe una base de datos online que describe las mutaciones halladas en POLG y los fenotipos asociados a ellas (http://tools.niehs.nih.gov/polg/).
Aspectos clínicos de las mutaciones en POLGPOLG y sus fenotiposDesde un punto de vista clínico, las mutaciones en POLG se pueden agrupar en formas de inicio infantil que incluyen: a) el síndrome de Alpers-Huttenlocher, y b) síndromes miohepatocerebrales de inicio infantil (MCHS, childhood myocerebrohepatopathy spectrum). Las formas de inicio más tardío incluyen: a) cuadros con afectación predominantemente ocular, con las llamadas oftalmoplejía crónica progresiva autosómica dominante o recesiva (adCPEO o arCPEO, autosomal dominant or recessive chronic progressive external ophtalmoplegia); b) cuadros de ataxia-neuropatía, que incluyen el síndrome de ataxia recesiva mitocondrial (MIRAS, mitochondrial recessive ataxia syndrome) y la ataxia sensorial, neuropatía, disartria y oftalmoplejía (SANDO, sensory ataxic neuropathy with dysarthria and opthalmoparesis), y c) síndromes combinados a otros cuadros (ataxia sensorial y elementos clínicos de la entidad MERRF (myoclonic encephalopathy and ragged red fibers) acuñado MEMSA (myoclonic epilepsy myopathy sensory ataxia) (tabla 1).
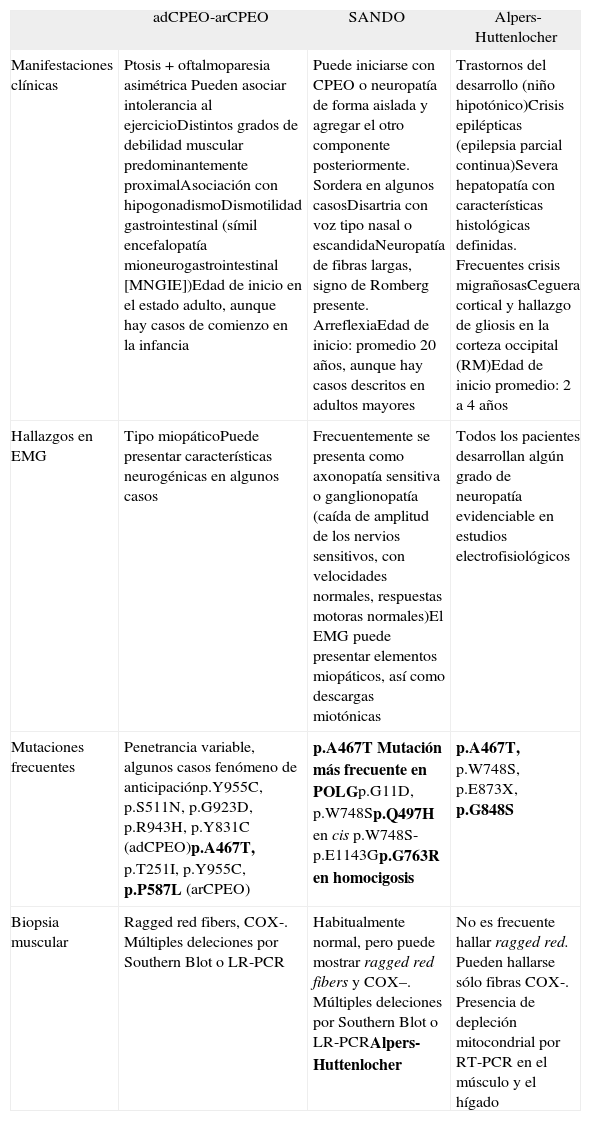
Cuadro esquemático que muestra los elementos característicos de las tres entidades clínicas más frecuentes asociadas a mutaciones en POLG
| adCPEO-arCPEO | SANDO | Alpers-Huttenlocher | |
| Manifestaciones clínicas | Ptosis + oftalmoparesia asimétrica Pueden asociar intolerancia al ejercicioDistintos grados de debilidad muscular predominantemente proximalAsociación con hipogonadismoDismotilidad gastrointestinal (símil encefalopatía mioneurogastrointestinal [MNGIE])Edad de inicio en el estado adulto, aunque hay casos de comienzo en la infancia | Puede iniciarse con CPEO o neuropatía de forma aislada y agregar el otro componente posteriormente. Sordera en algunos casosDisartria con voz tipo nasal o escandidaNeuropatía de fibras largas, signo de Romberg presente. ArreflexiaEdad de inicio: promedio 20 años, aunque hay casos descritos en adultos mayores | Trastornos del desarrollo (niño hipotónico)Crisis epilépticas (epilepsia parcial continua)Severa hepatopatía con características histológicas definidas. Frecuentes crisis migrañosasCeguera cortical y hallazgo de gliosis en la corteza occipital (RM)Edad de inicio promedio: 2 a 4 años |
| Hallazgos en EMG | Tipo miopáticoPuede presentar características neurogénicas en algunos casos | Frecuentemente se presenta como axonopatía sensitiva o ganglionopatía (caída de amplitud de los nervios sensitivos, con velocidades normales, respuestas motoras normales)El EMG puede presentar elementos miopáticos, así como descargas miotónicas | Todos los pacientes desarrollan algún grado de neuropatía evidenciable en estudios electrofisiológicos |
| Mutaciones frecuentes | Penetrancia variable, algunos casos fenómeno de anticipaciónp.Y955C, p.S511N, p.G923D, p.R943H, p.Y831C (adCPEO)p.A467T, p.T251I, p.Y955C, p.P587L (arCPEO) | p.A467T Mutación más frecuente en POLGp.G11D, p.W748Sp.Q497H en cis p.W748S-p.E1143Gp.G763R en homocigosis | p.A467T, p.W748S, p.E873X, p.G848S |
| Biopsia muscular | Ragged red fibers, COX-. Múltiples deleciones por Southern Blot o LR-PCR | Habitualmente normal, pero puede mostrar ragged red fibers y COX–. Múltiples deleciones por Southern Blot o LR-PCRAlpers-Huttenlocher | No es frecuente hallar ragged red. Pueden hallarse sólo fibras COX-. Presencia de depleción mitocondrial por RT-PCR en el músculo y el hígado |
LR-PCR: long range PCR; RT-PCR: real time PCR.
El síndrome de Alpers-Huttenlocher representa, en su forma completa, el extremo de severidad del espectro clínico correspondiente a mutaciones en POLG (OMIM#203700). Es una entidad clínica caracterizada por el compromiso hepatocerebral donde la forma de inicio más frecuente es la infantil (2 a 4 años)27-28, aunque se han descrito casos de inicio en el estado adulto29–31. Las crisis epilépticas son la manifestación inicial en el 50% de los pacientes. Pueden presentarse como convulsiones refractarias, habitualmente de inicio focal, que evolucionan a epilepsia parcial continua32,33. El retraso psicomotriz, las crisis migrañosas y la ceguera cortical son otros elementos frecuentes de hallar. El compromiso hepático es típicamente desencadenado tras el tratamiento con ácido valproico, en el intento de controlar las crisis epilépticas34–36. La biopsia presenta rasgos histológicos característicos que permiten diferenciarla de las producidas por causas tóxicas o farmacológicas34,37,38.
Los pacientes se pueden presentar desde el nacimiento con dificultades en el desarrollo psicomotor, un desarrollo normal con posterior evolución subaguda a algún gatillo ambiental o desarrollo normal con posterior pérdida de las pautas psicomotrices ya adquiridas. No es infrecuente hallar una severa neuropatía, coreoatetosis o incluso ceguera cortical como síntomas agregados. Los estudios de imágenes (RM) pueden mostrar compromiso cortical posterior simétrico (lóbulo occipital) en el cerebelo y el tálamo, a diferencia del síndrome de Leigh, en el que las lesiones se localizan con mayor frecuencia en los ganglios basales y en el tronco encefálico39. Las mutaciones más frecuentemente halladas son A467T y W748S31,33. No es habitual el hallazgo de fibras rojas rasgadas en la biopsia muscular, pero sí la presencia de depleción mitocondrial (disminución en el número de copias de ADNmt) por técnicas moleculares como la Real Time PCR (RT-PCR). El hallazgo de múltiples deleciones es menos frecuente, e inclusive en estadios precoces de la enfermedad ambas condiciones pueden estar ausentes.
Síndromes miohepatocerebrales de inicio infantil (MCHS)(MCHS) Incluyen un espectro de casos con inicio entre los primeros meses de vida y los tres años que incluye retraso psicomotriz o pérdida de pautas del desarrollo, acidosis láctica y miopatía con retraso en el crecimiento. A diferencia del síndrome de Alpers, no presentan los hallazgos histopatológicos característicos en la biopsia hepática y pueden no presentar convulsiones en estadios iniciales. Otros síntomas que pueden agregarse son vómitos cíclicos, hipoacusia, acidosis tubular renal, pancreatitis e insuficiencia hepática40.
adCPEO y arCPEOLa oftalmoparesia es un elemento predominante en la patología mitocondrial, pudiendo seguir distintos patrones de herencia. Los casos esporádicos habitualmente son producidos por grandes deleciones en el ADNmt41, mientras que las formas heredadas vía materna son debidos a mutaciones42 o deleciones puntuales del ADNmt (véase www.mitomap.org). A pesar de ello, se han descrito formas con un patrón de tipo mendeliano, ya sea autosómico recesivo (arCPEO)43 o dominante (adCPEO)44,45. Mutaciones en POLG1 constituyen el 45% de las formas mendelianas de CPEO. Otros genes descritos son PEO1 y ANT143,46. Estas formas por mutaciones nucleares con patrón recesivo se asocian habitualmente a un mayor compromiso multisistémico, especialmente la afectación cardiaca, la cual puede ser un elemento dominante, a diferencia de las formas esporádicas, donde los síntomas se restringen al compromiso ocular. La oftalmoparesia puede constituir un elemento más dentro de la encefalopatía mioneurogastrointestinal (MNGIE, myoneurogastrointestinal encephalopathy)47,48 o estar asociado a cuadros de parkinsonismo49, no así con la enfermedad de Parkinson idiopática50. En otros casos, la ataxia sensitiva puede preceder en años la aparición de la oftalmoparesia, evolucionando a un cuadro compatible con el síndrome SANDO (véase más adelante).
Habitualmente los cuadros clínicos de adCPEO se presentan con un curso relativamente más benigno que los descritos, aunque existe marcada variabilidad en su penetrancia, lo que podría estar vinculado con el tipo de mutación patogénica. Tal es el caso de Y955C, pues se ha visto que posee penetrancia completa con un fenotipo más severo, a diferencia de G923D, A957S, donde la expresión clínica es más variable y leve43. Además de la ptosis, puede observarse en estos pacientes compromiso de la deglución, debilidad muscular en las extremidades y cuadros severos de depresión.
En ambos casos es frecuente el hallazgo de fibras rojas rasgadas, COX negativas y múltiples deleciones a través de la técnica de Southern Blot.
Síndromes de ataxia sensitiva-neuropatíaDentro de este grupo se han incluido dos fenotipos en los cuales la manifestación predominante es la ataxia de la marcha producida por una severa neuropatía sensitiva y/o ganglionopatía, por el compromiso cerebeloso o una combinación de los previos. La ataxia sensorial producida por el compromiso neuropático y del ganglio de la raíz dorsal puede preceder en años a la instalación de las manifestaciones oculares (ptosis-oftalmoparesia)51. Si a ello se agrega disartria, conforma el síndrome SANDO. En 1997 Fadic et al52 realizaron la primera descripción de 4 pacientes con la tríada descrita y el hallazgo de múltiples deleciones en músculo y nervio. Clínicamente la neuropatía atáxica se debe a una severa disminución de la batiestesia y palestesia asociada al signo de Romberg y pérdida de reflejos miotáticos53. Algunos pacientes pueden presentar debilidad muscular sobreagregada.
La biopsia de nervio puede mostrar en estos casos pérdida mixta de fibras finas no mielinizadas y gruesas mielinizadas. La RM de encéfalo ocasionalmente presenta lesiones hiperintensas en T2 a nivel talámico bilateral54. Existe gran variabilidad en la edad de presentación del síndrome SANDO, con algunos casos que inician sus síntomas más allá de los 70 años55.
El síndrome MIRAS es una entidad inusual vinculada con mutaciones en POLG (W748S en cis con E1143G), la cual fue descrita inicialmente en pacientes finlandeses56. Consiste en una forma de ataxia recesiva que clínicamente combina el compromiso neuropático y lesiones en cerebelo sin oftalmoparesia, haciéndola en algunos casos indistinguible de las ataxias espinocerebelosas (SCA), aunque la edad de presentación es más tardía (tabla 2).
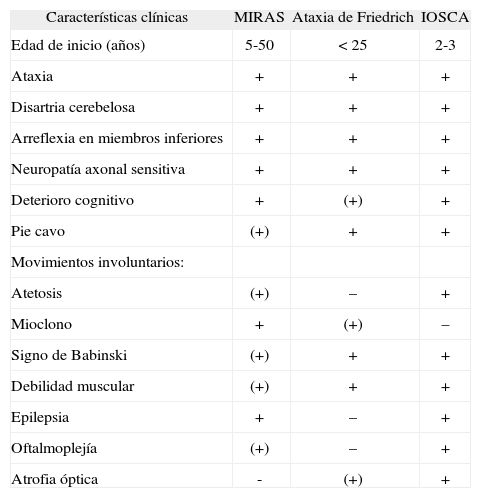
Características clínicas de las ataxias recesivas
| Características clínicas | MIRAS | Ataxia de Friedrich | IOSCA |
| Edad de inicio (años) | 5-50 | <25 | 2-3 |
| Ataxia | + | + | + |
| Disartria cerebelosa | + | + | + |
| Arreflexia en miembros inferiores | + | + | + |
| Neuropatía axonal sensitiva | + | + | + |
| Deterioro cognitivo | + | (+) | + |
| Pie cavo | (+) | + | + |
| Movimientos involuntarios: | |||
| Atetosis | (+) | – | + |
| Mioclono | + | (+) | – |
| Signo de Babinski | (+) | + | + |
| Debilidad muscular | (+) | + | + |
| Epilepsia | + | – | + |
| Oftalmoplejía | (+) | – | + |
| Atrofia óptica | - | (+) | + |
MIRAS: mitochondrial recessive ataxic syndrome; IOSCA: infantile onset spinocerebellar ataxia.
Es de destacar que el cambio de aminoácido p.E1143G es considerado habitualmente un polimorfismo (~3% de la población caucásica), aunque en MIRAS se encontró siempre presente en asociación con p.W748S. Para establecer el efecto de este polimorfismo sobre otras mutaciones son necesarios estudios funcionales.
Asimismo, existen reportes aislados de casos los cuales describen mutaciones en POLG vinculados a otras manifestaciones. Harrower et al57 describieron un paciente con un cuadro típico de Charcot Marie Tooth tipo 2 (CMT2) y la presencia de tres mutaciones en POLG1 (c.191C>T en el exón 2, c.695G>A en el exón 3 y c.2209G>C en el exón 13), apoyando la idea de la variabilidad en la expresión fenotípica de las mutaciones en este gen.
Van Goethem et al58 describieron un paciente con ataxia sensorial y elementos clínicos del MERRF aunque sin el hallazgo típico de fibras rojas rasgadas en las fibras musculares ni de ptosis palpebral. Este fenotipo abarcaría a las previamente nombradas ataxias espinocerebelosas con epilepsia (SCAE, spinocerebellar ataxia with epilepsy) y el síndrome MIRAS.
Recomendaciones respecto al diagnósticoFrente al desafío que impone para el médico la sospecha de encontrarse frente a una enfermedad mitocondrial, existen ciertos elementos que pueden ser tenidos en cuenta a fin de orientar el estudio genético en búsqueda de mutaciones en POLG1.
- •
En los pacientes con ptosis palpebral y oftalmoplejía que presentan agregación familiar con un patrón autosómico dominante o recesivo debería sospecharse en primera instancia una mutación en POLG1.
- •
En caso de ser negativo el estudio de este gen, debería considerarse la secuenciación de otros genes, como PEO1, ANT1 o incluso OPA1.
- •
En formas de CPEO esporádicas donde no se hallaron deleciones únicas o mutaciones puntuales en el ADNmt (habitualmente ARNt) debería secuenciarse POLG1 a fin de descartar mutaciones nucleares.
- •
La asociación de una severa neuropatía axonal mixta, ptosis - oftalmoplejía más diferentes grados de disartria, debe hacer sospechar un síndrome SANDO y, por ende, descartar mutaciones en POLG1.
- •
Mutaciones en POLG1 deberían incluirse como parte del cribado genético de ataxias recesivas.
- •
Mutaciones en POLG1 frecuentemente se asocian al hallazgo de múltiples deleciones por técnicas moleculares como Southern Blot o Long Range PCR, por lo que estas técnicas, junto con la clínica, pueden orientar el análisis de este gen.
- •
La ausencia de fibras rojas rasgadas en la biopsia muscular no descarta el diagnóstico, más aún en los casos pediátricos donde su hallazgo es infrecuente.
- •
Se recomienda la secuenciación del gen de forma completa, ya que —como se ha mencionado— no existe una correlación geno-fenotipíca estrecha y la distribución de las mutaciones no está restringida a una región específica de gen.
- •
La tríada de encefalopatía hepática severa desencadenada tras un evento agudo como un síndrome febril o la introducción del ácido valproico, crisis epilépticas de inicio parcial y retardo psicomotor o pérdida de pautas de desarrollo debe sugerir SAH. El hallazgo de depleción mitocondrial por estudios genéticos apoya el diagnóstico, aunque se han descrito otros genes (DGUOK y MPV17).
POLG γ es una de las enzimas que conforman el aparato de replicación mitocondrial, y sus componentes están codificados por los genes POLG1 y POLG2. Las mutaciones del gen POLG1 constituyen una causa frecuente de defectos en el funcionamiento mitocondrial, abarcando un espectro de diversos fenotipos no descritos previamente o la reclasificación de otras entidades en síndromes comunes (SCA). Además, existen escasos reportes de mutaciones en POLG2, que codifican para las subunidades accesorias de la enzima. A pesar de la frecuencia de defectos en POLG1, no es posible establecer por el momento una clasificación desde un punto de vista genético-estructural ni cuál es el rol funcional de algunas sustituciones encontradas en el gen y la interacción de las mismas junto a otras mutaciones. Mutaciones en POLG1 deberían ser consideradas en pacientes con adCPEO, arCPEO, ataxia-neuropatía o síndrome de Alpers tras el hallazgo de múltiples deleciones o depleción mitocondrial, mediante técnicas moleculares como el Southern Blot o la Long Range PCR.
Financiación y Conflicto de interesesNo se recibió financiación para la realización del presente trabajo. No existen conflictos de intereses.
AgradecimientosSe agradece a los miembros de la sección de Enfermedades Neuromusculares del Hospital Británico por la corrección del manuscrito.
