





La presentación clínica del síndrome de Guillain-Barré puede ser heterogénea debido a sus múltiples variantes, algunas definidas y otras que forman parte del espectro de la enfermedad.
Caso clínicoMujer de 35 años que presentó neuropatía craneal múltiple, sin otros hallazgos. Se realizó determinación de anticuerpos antigangliósidos en el líquido cefalorraquídeo, positivos para GD1b y GQ1b. Se pautó tratamiento con gammaglobulina con buena respuesta.
ConclusiónLa neuropatía craneal múltiple puede ocurrir en el contexto de variantes de Guillain-Barré. Con relación al caso presentado, la distribución de los anticuerpos GD1b y GQ1b explicaría la presentación clínica de la paciente.
The clinical presentation of Guillain-Barré syndrome can be heterogeneous due to its multiple variants, some defined and others that are part of the spectrum of the disease.
Clinical caseThirty-five-year-old female who presented with multiple cranial neuropathy, with no other findings. Determination of antiganglioside antibodies in cerebrospinal fluid, positive for GD1b and GQ1b, was performed. Treatment with gamma globulin was performed with good response.
ConclusionMultiple cranial neuropathy can occur in the context of Guillain-Barré variants, in relation to the case presented, the distribution of GD1b and GQ1b antibodies would explain the clinical presentation of the patient.
El síndrome de Guillain-Barré es una polineuropatía aguda de carácter autoinmune que se presenta como una enfermedad paralizante monofásica aguda. En la mayoría de los casos, se ve desencadenada por una infección previa, ya que es causada por una activación inmune aberrante, con daño consiguiente de los nervios periféricos, aunque su fisiopatología permanece aún sin dilucidar por completo.
Fue descrito por primera vez en el año 1916 por 3neurólogos franceses durante la primera guerra mundial con relación a 2soldados que presentaron parálisis ascendente y transitoria: Georges Guillain, Jean-Alexandre Barré y André Strohl. El término fue utilizado por primera vez en el año 1927. Se desconocen las razones de la omisión de Strohl.
Es la polineuropatía aguda más frecuente del mundo occidental, con una incidencia de 2 casos cada 100.000 habitantes al año, con mayor frecuencia en varones y una incidencia que disminuye con la edad1.
La presentación clínica típica consiste en debilidad ascendente que comienza en los miembros inferiores para luego afectar a los miembros superiores, músculos axiales y craneales. No obstante, la presentación clínica de la enfermedad puede ser heterogénea, ya que se han identificado múltiples variantes.
Presentamos el caso de una paciente con afectación aguda y progresiva de múltiples pares craneales y parestesias en ambos miembros superiores, sin ataxia ni debilidad de las extremidades, con reflejos osteotendinosos normales.
Caso clínicoPaciente femenina de 35 años de edad sin antecedentes de relevancia que consultó al servicio de urgencias por diplopía binocular de 48 h de evolución. Al interrogatorio dirigido refirió diagnóstico de otitis externa 10 días antes de la consulta actual.
Al examen físico neurológico se constató inicialmente, como hallazgo positivo, la presencia de diplopía binocular a mirada primaria, que empeoraba al mirar hacia los laterales, sin compromiso evidenciable en el examen físico de afectación de los pares craneales. A las 24 h presentó restricción de la abducción bilateral, compatible con paresia de ambos rectos externos inervados por el vi par craneal. El examen de fuerza muscular, sensibilidad y reflejos osteotendinosos fue normal (2/4 en los 4miembros).
Se decidió internación para estudio de parálisis bilateral del vi par craneal.
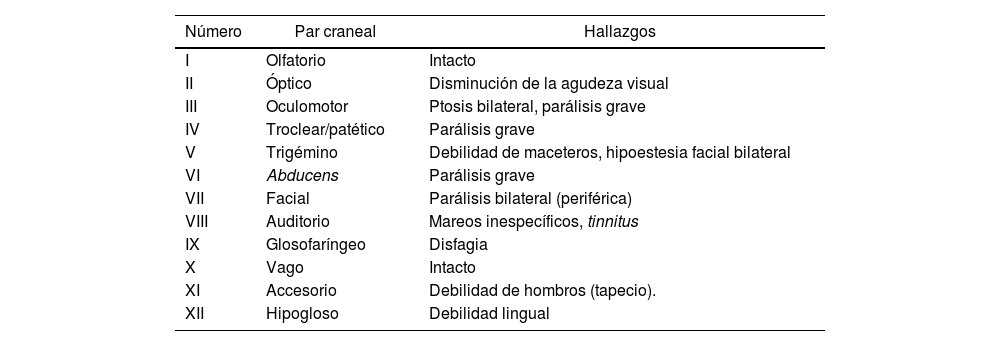
A las 48 h de iniciados los síntomas, la paciente evolucionó con ptosis palpebral bilateral, oftalmoplejía bilateral y parestesias en ambos miembros superiores. A las 4 días de iniciados los síntomas, se agregó diplejía facial y debilidad de lengua, cuello y hombros. Se solicitó pase a unidad de cuidados intensivos para monitoreo neurológico (fig. 1 y tabla 1).
 representando la evolución de los síntomas presentados por la paciente.")
| Número | Par craneal | Hallazgos |
|---|---|---|
| I | Olfatorio | Intacto |
| II | Óptico | Disminución de la agudeza visual |
| III | Oculomotor | Ptosis bilateral, parálisis grave |
| IV | Troclear/patético | Parálisis grave |
| V | Trigémino | Debilidad de maceteros, hipoestesia facial bilateral |
| VI | Abducens | Parálisis grave |
| VII | Facial | Parálisis bilateral (periférica) |
| VIII | Auditorio | Mareos inespecíficos, tinnitus |
| IX | Glosofaríngeo | Disfagia |
| X | Vago | Intacto |
| XI | Accesorio | Debilidad de hombros (tapecio). |
| XII | Hipogloso | Debilidad lingual |
Se realizó resonancia magnética de cerebro con contraste sin evidencia de lesiones, punción lumbar a las 72 h de iniciados los síntomas sin disociación de albúmina citológica (34 proteínas, 3 leucocitos), con serología bacteriana y PCR virales negativas.
La electromiografía de fibra única y la estimulación repetitiva fueron normales.
En el fondo de ojos no se observaron particularidades y en la resonancia de órbitas con contraste no hubo evidencia de lesiones.
Debido a la evolución clínica de la paciente y los estudios complementarios, se sospechó una variante de síndrome de Guillain-Barré, por lo que se extrajo suero para la realización de panel de antigangliósidos y, posteriormente, se inició tratamiento inmunomodulador con gammaglobulina humana a razón de 2 g/kg durante un total de 5 días.
La paciente fue dada de alta tras un periodo de observación en el que se constató estabilidad clínica, sin evidencia de debilidad bulbar pero con persistencia de la diplejía facial y la oftalmoplejía bilateral. Los reflejos osteotendinosos se mantuvieron normales en toda la evolución clínica.
Posteriormente, se recibieron los resultados de panel de antigangliósidos, que eran positivos para GD1b (razón 148) y GQ1b (razón 160), por lo que se interpretó el cuadro clínico como un espectro clínico del síndrome de Guillain-Barré seropositivo para ambos anticuerpos.
Luego de 45 días de evolución del cuadro clínico, la paciente acudió a control, en el que se evidenció mejoría del cuadro clínico, con mayor fuerza facial bilateral y presencia de movimientos oculares que le generaban diplopía. No presentó nuevos episodios de debilidad bulbar.
DiscusiónSe ha descrito en la literatura médica una gran variedad de presentaciones clínicas sobre las neuropatías inflamatorias agudas inmunomediadas, de las que el síndrome más característico es el de Guillain-Barré clásico, caracterizado por hiporreflexia y alteración de la sensibilidad y fuerza muscular ascendentes.
En cuanto a sus variantes, se encuentran la forma paraparética, sensitiva pura, motora pura, debilidad faríngea-cervical-braquial, parálisis facial con parestesias, el síndrome de Miller Fisher (SMF) y la encefalitis de Bickerstaff2. Algunas de estas suelen tener presentaciones atípicas, incluyendo la neuropatía craneal múltiple3,4, o superponerse entre ellas, especialmente aquellas con características de SMF, SGB y de la encefalitis de Bickerstaff, debido a que forman parte del espectro de los síndromes anti-GQ1b2,5,6. La paciente presentada no cumplía criterios para ninguna de estas variantes.
Tras varias investigaciones, se ha propuesto a los anticuerpos antigangliósidos como los responsables de las neuropatías inmunomediadas. La porción de ácido siálico presente en la secuencia del hidrato de carbono, denominados GM1, GD1a, GD1b, GQ1b y GT1b, tiene una distribución relativa única dentro del sistema nervioso periférico, por lo tanto, los anticuerpos antigangliósidos pueden unirse a estos epítopos extracelulares para inducir lesión de la membrana y presentar diferente sintomatología de acuerdo con su localización7. Los gangliósidos pueden unirse y formar grupos en la membrana celular. Se ha visto en algunas formas de SGB y SMF presencia de anticuerpos contra complejos de gangliósidos, como GM1/GD1a, GM1/GT1b, GQ1b/GD1b, GQ1b/GD1a, entre otros5,7,8.
La neuropatía craneal múltiple puede ocurrir en el contexto de variantes del síndrome de Guillain-Barré, que representa alrededor del 3 al 5% de los casos. En la mayoría de ellos, la afectación se limita casi exclusivamente a los pares craneales VII, IX y X lo que se traduce en parálisis facial, disartria y disfagia9. Se ha descrito que la limitación de la afectación a los pares craneales guarda relación con la localización de gangliósidos GQ1b, la cual abunda en los pares craneales oculomotores (III, IV, VI) además del II par craneal con relación a otros nervios craneales. De la misma manera, se encontró que el porcentaje de GQ1b fue mayor en el V par craneal con relación a los demás anticuerpos antigangliósidos. Respecto al caso presentado, se sabe, además, que los antigangliósidos también están representados en los pares craneales III, IV y VI10, aunque esta disposición no es suficiente para explicar los hallazgos en este caso, por lo que otros factores, como la presencia de anticuerpos GD1b, puede ser causa de la presentación clínica. En la investigación ya citada se encontró que los anticuerpos GD3, GD1b y GT1b fueron los componentes principales de todos los nervios craneales, exceptuando el porcentaje de GD1b, que fue menor en el nervio XI.
Ante la presencia de diplopía, oftalmoplejía extraocular y diplejía facial simétrica de presentación aguda, se consideraron varios diagnósticos diferenciales con la finalidad de brindar tratamiento acorde. Entre las posibles causas se tomaron en cuenta las vasculares, tumorales, traumáticas, infecciosas, la esclerosis múltiple, diabetes mellitus, enfermedad tiroidea, otras autoinmunes, déficit de vitaminas y miastenia gravis11, que se descartaron al presentar perfiles virales y bacteriológicos negativos tanto en el suero como en el líquido cefalorraquídeo, virus de la inmunodeficiencia humana y VDRL negativos así como los anticuerpos para enfermedades sistémicas, anticuerpos anti-Musk y ACRA y estudios electrofisiológicos, como electromiograma de fibra única y estimulación repetitiva.
La particularidad del caso reportado radica en la mayor afectación de los pares craneales, ya que afectaba casi a su totalidad (exceptuando pares I y X).
ConclusionesAnte la presencia de un paciente con neuropatía craneal múltiple de rápida progresión deben descartarse una serie de diagnósticos diferenciales dentro de los cuales se deben tener en cuenta las variantes del síndrome de Guillain-Barré, para lo que se debe tener un alto nivel de sospecha, aun cuando las características del paciente no se encasillen en las variantes descritas, debido a la gran expresión clínica del espectro de los anticuerpos antigangliósidos.
FinanciaciónNinguna.
Conflicto de interesesNinguno para declarar.
