




Pathogenic variants in STUB1 can be causal under dominant (SCA48) and recessive (SCAR16) inheritance patterns. We report a SCA48 patient with ataxia, dementia and movement disorders.
Clinical caseWe describe a 45 year-old man with cognitive impairment, ataxia, bradykinesia, tremor and chorea. A NGS-based multigene panel revealed the presence of a novel heterozygous likely pathogenic variant in STUB1 gene confirming a diagnosis of SCA48.
ConclusionsHyper and hypokinetic movement disorders seems to be a hallmark of SCA48. The frequent association of ataxia, chorea and cognitive impairment suggest consider SCA48 as a differential diagnosis of SCA17 and other Huntington-like disorders.
Variantes patogénicas en el gen STUB1 pueden manifestarse bajo un patrón autosómico dominante (SCA48) o recesivo (SCAR16) de herencia. Reportamos un paciente con SCA48 con ataxia, demencia y movimientos anormales.
Caso clínicoDescribimos el caso de un varón de 45 años con compromiso cognitivo, ataxia, bradicinesia, temblor y corea. Mediante un panel de ataxias se evidenció la presencia de una variante nueva probablemente patogénica en el gen STUB1, confirmando el diagnóstico de SCA48.
ConclusionesTrastornos del movimiento hipo e hipercinéticos demuestran ser una característica de los pacientes con SCA48. La asociación frecuente de ataxia, compromiso cognitivo y corea lleva a considerar SCA48 como diagnóstico diferencial de SCA17 y otras entidades similares a Huntington.
Biallelic mutations in the STUB1 gene, that encodes the E3 ubiquitin ligase CHIP, are the cause of SCAR16. An early-onset recessive ataxia with a wide range of cerebellar and extra-cerebellar symptoms and occasionally hypogonadism.1,2 Moreover, dominant ataxia families segregating heterozygous mutations in STUB1 have been recently described.3 Thus, pathogenic variants in STUB1 can be causal under dominant (SCA48) and recessive (SCAR16) inheritance patterns.4 SCA48 patients often present with extra-cerebellar manifestations that include movement disorders and cognitive decline. Here, we report on a new SCA48 patient that presented with ataxia, parkinsonism, cognitive impairment and chorea along with a review of the movement disorders previously described in SCA48 patients.
Clinical caseThis 45 year-old man was referred to our Neurogenetics Unit due to the presence of cognitive impairment and ataxia. He was apparently healthy until 6 months previous to our evaluation, when he started with progressive memory loss, apathy, dysarthria and limb incoordination. His father, who died at the age of 65, was diagnosed with schizophrenia. His unique sister at the age of 30 started suffering from a progressive disorder characterized by the presence of cognitive impairment and involuntary movements. She died at the age of 50 without a definitive diagnosis. Neurological examination was remarkable for the presence of hypometric saccadic ocular movements, mild dysarthria and generalized hyporeflexia without gross abnormalities in sensation. He showed mild axial and appendicular ataxia (SARA scale of 9 points) and asymmetric bradykinesia which was more pronounced in the right side. Mild postural bilateral tremor and chorea were also evident in both upper limbs (Video 1). Neuropsychological evaluation documented multidomain cognitive impairment with failures in logic memory, attention, executive functions and verbal fluency. The MRI showed mild cortical and cerebellar atrophy and T2WI hyperintensity of dentate nuclei, the so-called “crab sign”. The patient tested negative for abnormal repeat expansions in the SCA1, SCA2, SCA3 and SCA17 genes. A NGS-based multigene panel for hereditary ataxias revealed the presence of a novel heterozygous likely pathogenic variant in STUB1 gene (NM_005861.4:c.854C>A (p.Ala285Asp)), confirming a diagnosis of SCA48.
CommentsThe case reported here highlights the frequent presence of extra-cerebellar manifestations in SCA patients, being movement disorders the most common ones in about a third of them5 and distinctive enough in some cases to suggest specific conditions such as parkinsonism in SCA2 and chorea in SCA17.6 The fifteen SCA48 families described in the literature3,4,7–11 and a recent study that included a large series of 47 patients from 28 families with STUB1 mutations and autosomal dominant inheritance,12 have in common a complex phenotype characterized by motor and cognitive impairments.
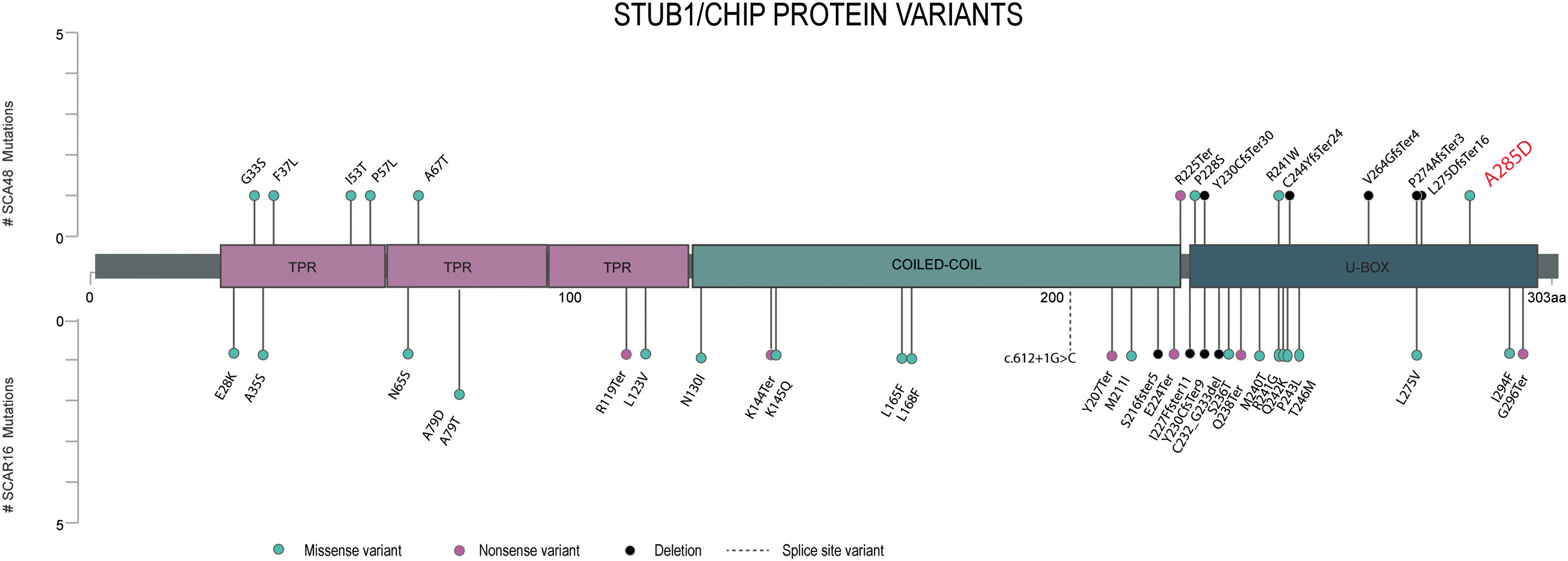
In SCA48 patients, different types of variants have been described and are distributed throughout the entire gene (Fig. 1), with the exception of the coiled-coil domain who influences the dimerization and stability of the whole protein. To date only one variant has been reported in SCA48 and SCAR16 (p. Y230CfsTer9)4 however, it has not yet been clarified how the type and location of STUB1 variants could be contributing to the differentiation of these two entities.

Lollipop plot displaying the position of the STUB1 pathogenic or likely pathogenic variants previously reported. On the horizontal axis we show the amino-acid position of each pathogenic variant identified until now. Variants associated with SCA48 are depicted by the upward lollipops and variants associated with SCAR16 are depicted by the downward lollipops. The variant detected in our patient is in red.
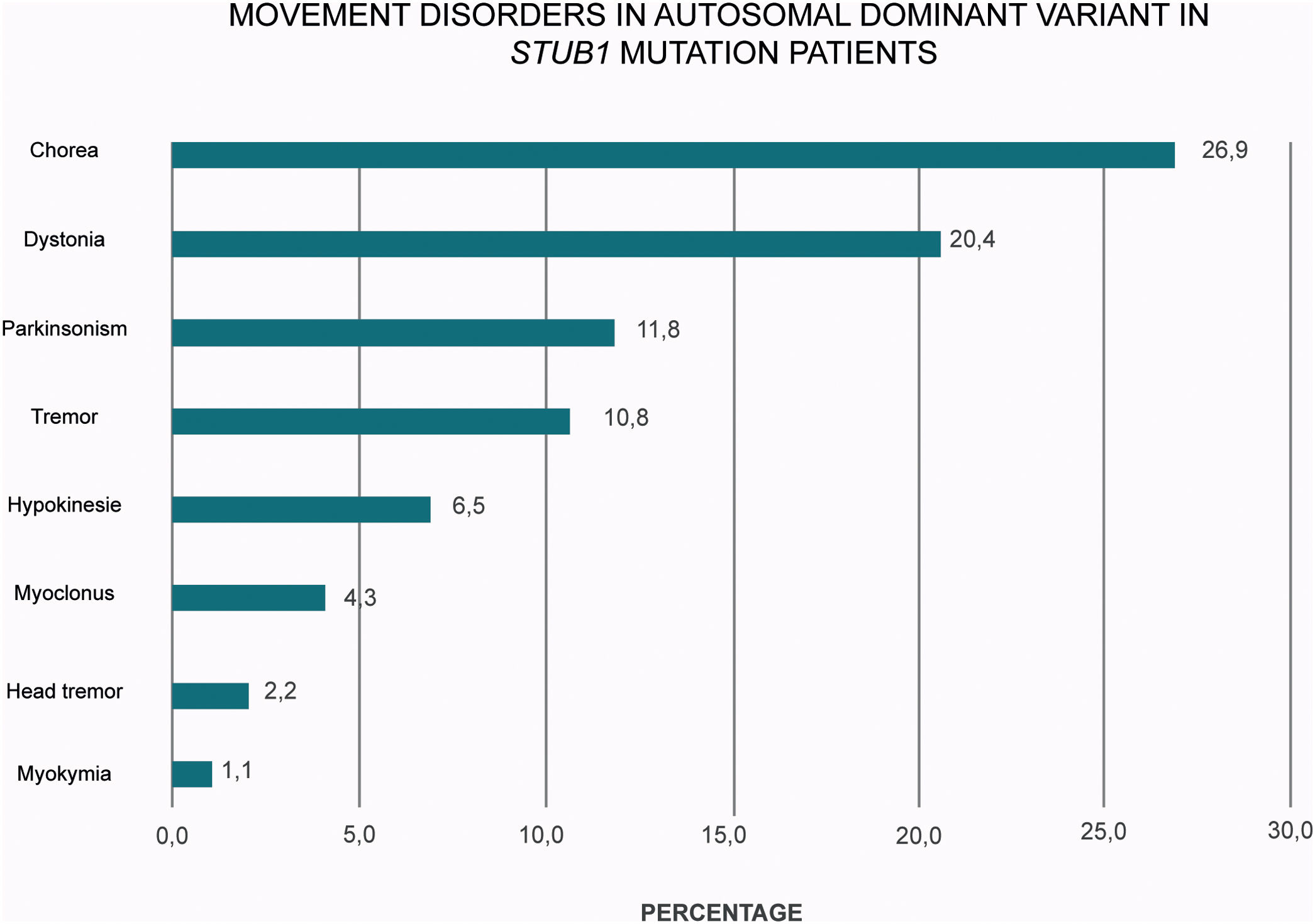
A plethora of hyper and hypokinetic movement disorders seems to be a hallmark of SCA48 (Table 1 and Fig. 2). Chorea is a particularly common feature (27% of the cases) followed by dystonia (20%) and parkinsonism (12%). Therefore, this frequent picture of ataxia, chorea and cognitive impairment in SCA48 patients make this condition to be considered as a mimic of SCA17 and other Huntington-like disorders.

SRQ: 1A, 1B, 1C, 3A, 3B.
JPM: 1A, 1C, 3A.
LZ: 1A, 1C, 3A.
MK: 1A, 1B, 1C, 3B, 3C.
1. Research project: A. Conception, B. Organization, C. Execution.
2. Statistical analysis: A. Design, B. Execution, C. Review and Critique.
3. Manuscript preparation: A. Writing of the first draft, B. Review and Critique, C. Final version approval.
Ethical compliance statementThis study was approved by the Institutional Ethics Committee of the Hospital JM Ramos Mejia of Buenos Aires, Argentina. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines. The authors confirm that the patient provided verbal and written consent for this work but because this article is a case report no IRB approval was necessary.
Financial disclosures for the previous 12 monthsJosefina Perez Maturo has received scholarship support from Argentinean National Science Council (CONICET). Marcelo Kauffman and Marcela Gonzalez Cid have received grant support from Ministry of Health of Buenos Aires City, Argentinean National Science Council (CONICET). Marcelo Kauffman has received grant support from Argentinean Ministry of Science and Technology and serves as Associate Editor of the journal Neurología Argentina. The rest of the authors declare that they have no conflict of interest.
Funding sourcesNo specific funding was received for this work.
Conflict of interestThe authors declare that there are no conflicts of interest relevant to this work.
The following are the supplementary data to this article:
This video shows main findings during neurological examination in the case reported. Segment 1 shows the presence of hypometric horizontal and vertical saccadic ocular movements. Segment 2–3 shows the presence of mild appendicular ataxia in upper limbs. Segment 5 shows the presence of bilateral postural tremor. Segment 6 shows bilateral asymmetric bradykinesia, more pronounced in the left side. Segment 7–9 shows ataxia in lower limbs and gait impairment predominant visible when walking or stand in tandem. Segment 10 shows the presence of bilateral distal mild choreic movements in upper limbs.
