





La neuromielitis óptica (NMO) es una enfermedad desmielinizante, autoinmune e inflamatoria crónica del sistema nervioso central caracterizada por ataques de neuritis óptica y mielitis longitudinal extensa (LETM). La presencia de un anticuerpo específico llamado IgG-NMO en suero ha sido de gran ayuda, tanto para la diferenciación con la esclerosis múltiple como para definir el espectro de la NMO. Este anticuerpo IgG1 actúa sobre los canales de agua (acuaporina 4) en el pie del astrocito. La seropositividad en el espectro de la NMO se relaciona con una probabilidad elevada de recaídas. Por lo tanto, la terapia preventiva con fármacos inmunosupresores debería ser instituida tan rápido como sea posible, dado que la discapacidad se relaciona con el efecto residual de los ataques. Se presenta el caso de una paciente con LETM que no cumple criterios para NMO definitiva ni para esclerosis múltiple donde el anticuerpo IgG-NMO positivo llevó a la decisión de iniciar tratamiento inmunosupresor. Se realiza una breve revisión del tema.
Neuromyelitis optica (NMO) is a demyelinating, autoimmune, and inflammatory disease of the central nervous system characterized by attacks of optic neuritis and transverse myelitis extensive. The presence of a specific antibody called NMO-IgG in serum has been of great help not only for differentiation with multiple sclerosis but to help define the spectrum of NMO. This IgG1 antibody acts on the water channels (aquaporin 4) at the foot of astrocyte. Seropositivity in the NMO spectrum relates to the high probability of relapses. Therefore, preventive therapy with immunosuppressant drugs should be instituted as early as possible because the disability is related to the residualeffect of the attacks. We report a case of a patient with extensive longitudinal myelopathy that does not meet criteria for definite NMO and multiple sclerosis where positive NMO-IgG antibody led to decision to initiate immunosuppressive therapy. We perform a brief review of the topic.
La NMO (o enfermedad de Devic) es una enfermedad desmielinizante, autoinmune e inflamatoria crónica del sistema nervioso central (SNC) que se caracteriza por afectar de forma severa a los nervios ópticos y la médula espinal, causando discapacidad en jóvenes y adultos1. Clínicamente se puede manifestar de forma monofásica o polifásica2. Durante décadas la NMO fue clasificada como una variante de la esclerosis múltiple (EM), fundamentalmente como la variante óptico espinal asiática (OSMS)1–3. Sin embargo, actualmente existen varias características que la distinguen de la EM3. En 2004 Lennon et al. reportaron la presencia de un autoanticuerpo IgG1 específico (91%) y sensible (73%) en suero que se une a los canales de agua diseminados en el SNC llamados acuaporina-4 (AQP4), el cual se expresa en los pies de los astrocitos2,4. Esto revolucionó el enfoque de la enfermedad, principalmente en el diagnóstico diferencial con la EM, dado que estos anticuerpos no estaban presentes en dicha enfermedad1–4. Asimismo, en el año 2006 y ante el descubrimiento de la IgG-NMO se revisaron los criterios de NMO y se amplió el espectro a formas clínicas limitadas (NMO spectrum disorders [NMOSD]) entre las que se encuentra la neuritis óptica (NO) y la mielitis longitudinal extensa (LETM) (lesión de 3 o más segmentos medulares) tanto aislada como recurrente, entre otras (tabla 1)1,2,5–7. Estos hallazgos tienen una implicación fundamental en la práctica clínica, principalmente para el inicio, tan precoz como sea posible, del tratamiento inmunosupresor8.
Desórdenes del espectro de la NMO
| Desórdenes del espectro de la NMO1 |
| 1. NMO |
| Formas limitadas de NMO |
| 2. Eventos «idiopáticos» simples o recurrentes de LETM en RMN |
| 3. NO recurrente o simultánea bilateral |
| 4. OSMS en asiáticos |
| 5. NO o LETM asociado a enfermedades autoinmunes sistémicas |
| 6. NO o LETM asociado con lesiones típicas de NMO (hipotálamo, cuerpo calloso, periventricular o tronco) |
| 7. Vómitos o hipo intratable7 |
| 8. Síndrome de encefalopatía posterior reversible7 |
| 9. Sintomatología de narcolepsia7 |
LETM: mielitis transversa longitudinal extensa; NMO: neuromielitis óptica; NO: neuritis óptica; OSMS: esclerosis múltiple óptico espinal; RMN: resonancia magnética nuclear.
Se presenta el caso de una paciente con LETM en el cual la presencia de IgG-NMO fue importante y fundamental para el diagnóstico y tratamiento. Además, realizaremos una breve revisión de la bibliografía internacional.
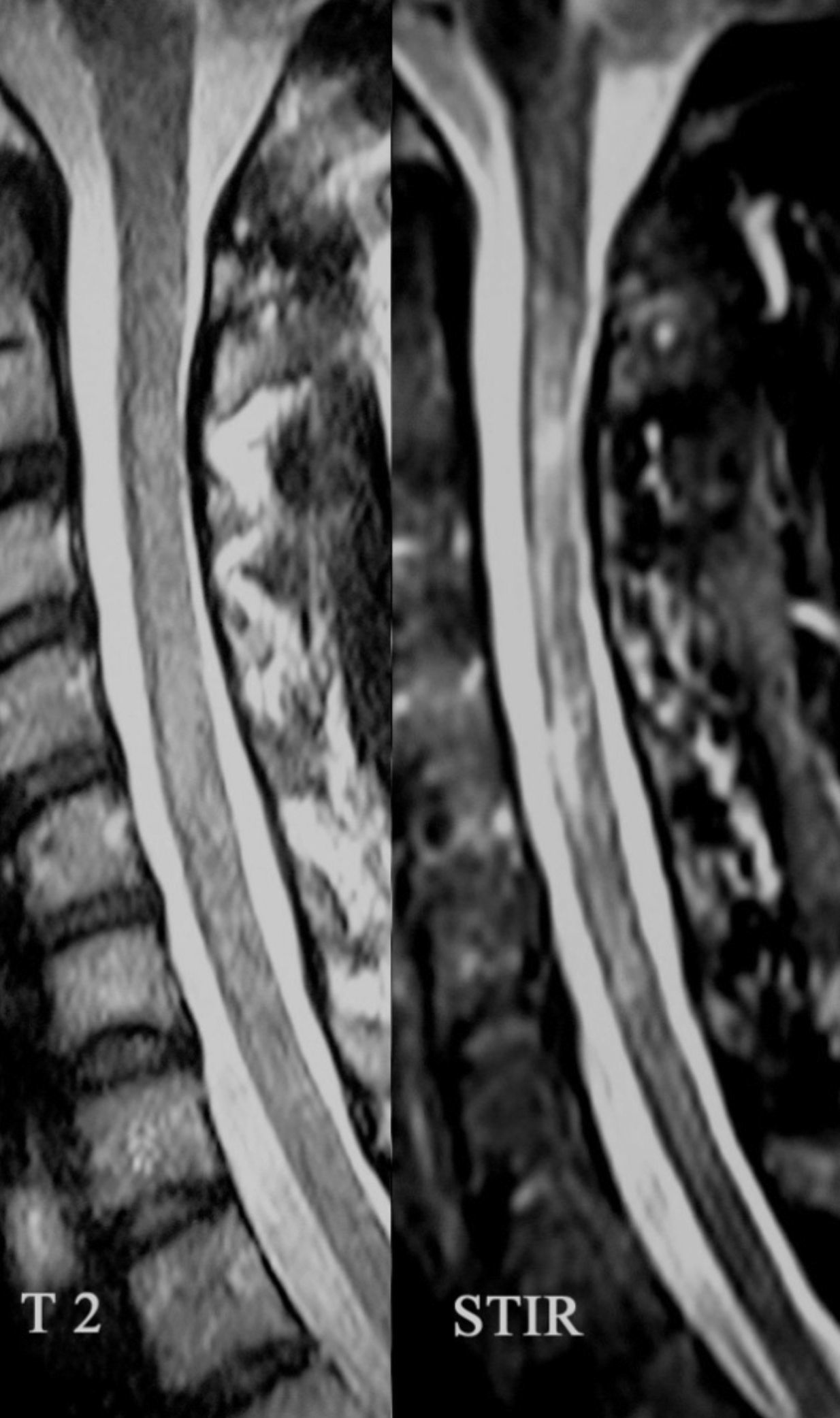
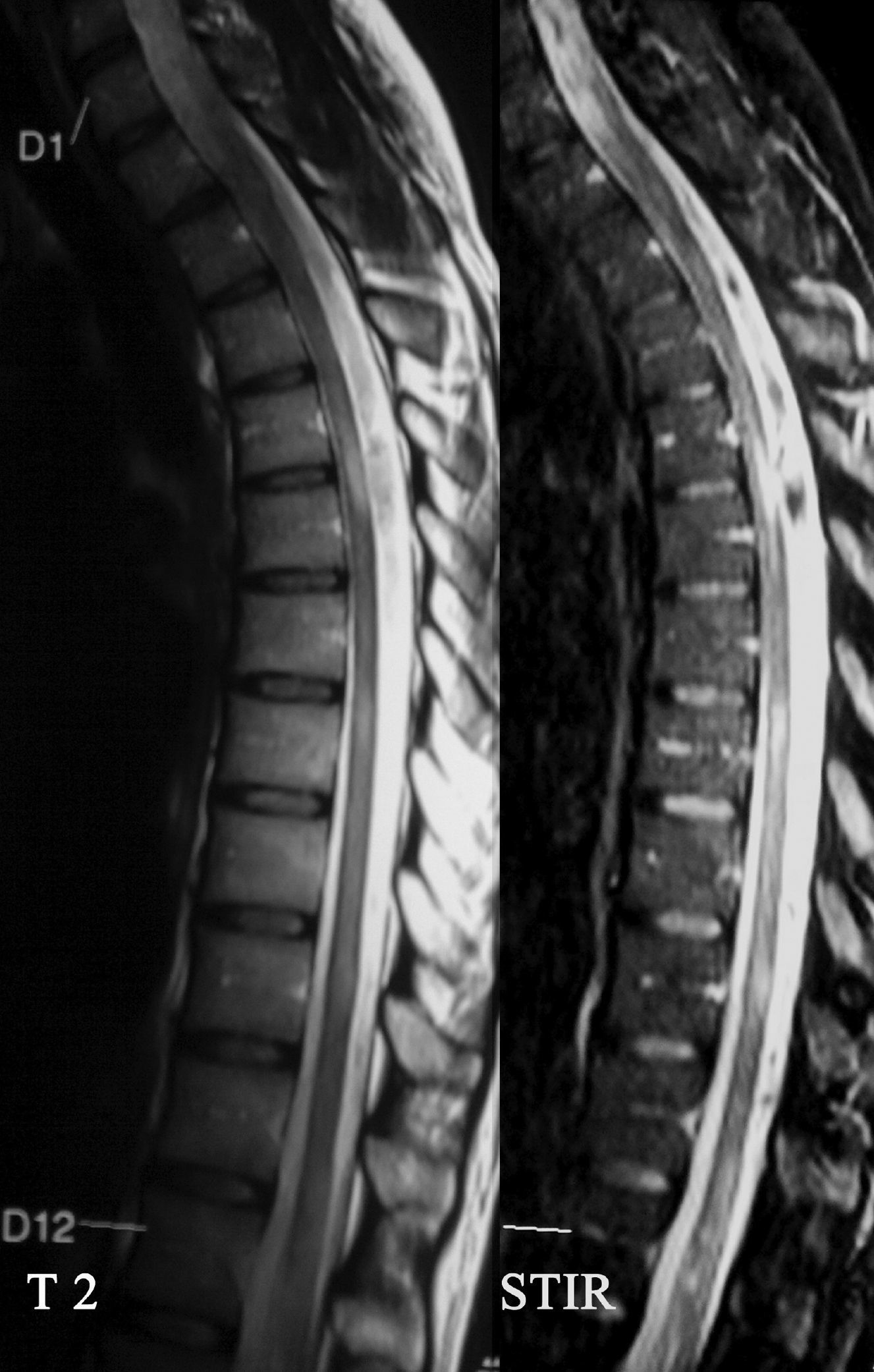
Caso clínicoMujer de 39 años con antecedentes de 2 abortos espontáneos, fenómeno de Raynaud y psoriasis leve. Consulta por dolor en el miembro inferior izquierdo, dificultad en la marcha y astenia de 2 años de evolución. En el examen físico se constata una leve paraparesia de predominio izquierdo, hipertonía, hiperreflexia y Babinski bilateral. La resonancia magnética (RM) de cerebro resultó normal; en la médula espinal se observan áreas focales hiperintensas en T2 y STIR a nivel cervical y dorsal (C2 a C5 parcheadas e irregulares, D5-D6, y más tenue a nivel D7-D8, D9-D10, y probablemente D11-D12) sin refuerzo con gadolinio. La RM a los 6 meses objetivó un cerebro normal y médula con persistencia de las lesiones, siendo más evidentes las lesiones a nivel D7 a D10 con un leve realce con gadolinio en D8 (figs. 1 y 2). Laboratorio: FAN+1/320 nuclear homogéneo, C3: 72 (84-193), C4: 6 (20-40),vitamina B12 normal, HIV, VDRL, hepatitis B, anti-Ro y anti-La, anti ADN, RNP, anticoagulante lúpico, anticardiolipinas y anti B2GP1 negativos. Función tiroidea normal con anti-TPO 314.96 (positivo). Líquido cefalorraquídeo (LCR) fisicoquímico y citológico normal, marcado fraccionamiento oligoclonal de IgG en el LCR con suero normal e índice de IgG de 10% (normal). Potencial evocado visual (flash y damero) normal. IgG-NMO positivo (método: inmunofluorescencia indirecta [IFI] confocal en corte de cerebelo de mono). Con diagnóstico de un síndrome incluido en el espectro de la NMO se inició azatioprina, llegando a una dosis de 100mg/día como tratamiento preventivo de próximas recaídas. Durante el seguimiento a 18 meses la paciente no presentó nuevas recaídas por clínica ni por imágenes.


Si bien la NMO es un enfermedad desmielinizante rara en pacientes caucásicos, su frecuencia sería mayor en otras poblaciones1,4,9. En Estados Unidos, Canadá y Europa constituye solo el 1,5-2% de las enfermedades desmielinizantes del SNC, mientras que en la población asiática e India llega a ser causa del 48% de estas enfermedades1,4. La prevalencia estimada es de 0,3-4,4 por 100.000 habitantes4. El pico de incidencia con respecto a la edad se encuentra en los 39 años, y en el sexo femenino es 9 veces más frecuente que en el masculino1. Aunque la susceptibilidad genética no está clara, existe una prevalencia aumentada en la población asiática, donde la OSMS/NMO se asoció a HLA-DPB1*0501 principalmente en pacientes con IgG-NMO positivo10. El descubrimiento del autoanticuerpo IgG-NMO en suero de pacientes con NMO fue realizado originalmente por IFI sobre un sustrato de tejido de SNC de ratón, observándose un patrón de inmunotinción específico que rodea la microvasculatura, pía, subpía y espacios de Virchow Robin1,4. Desde su descripción se han reportado múltiples estudios con diferentes métodos (IFI, radioinmunoprecipitación, Western Blot, ELISA y citometría de flujo, solos o en combinación) y distintos sustratos (tejido de SNC de ratón, rata, mono, así como células transfectadas y proteínas recombinantes de AQP4) presentando diferente sensibilidad y especificidad1–5. En un estudio reciente multicéntrico11 (EE. UU., Canadá e Inglaterra) se compararon 6 métodos diferentes para la determinación de IgG-NMO en pacientes con NMO (NMOSD: 25 y NMO: 35) vs controles (EM: 39, enfermedades autoinmunes: 25 y controles sanos: 22) midiendo la sensibilidad y especificidad del método. Los autores encontraron que la citometría de flujo cuantitativa y la observación de células transfectadas (CBA-E) obtuvieron la mejor sensibilidad, llegando al 77% 46 de 60 pacientes y al 73% 44 de 60 pacientes respectivamente. Asimismo, cuando combinaron CBA y ELISA se obtuvo un 100% de especificidad con una sensibilidad del 72% (43 de 60 pacientes). Es por ello que se recomienda como screening el uso de CBA-E para el diagnóstico y posterior tratamiento temprano en el espectro de la NMO.
Los anticuerpos IgG-NMO son patogénicos, y debido a que son del tipo IgG1 provocan citotoxicidad dependiente del complemento3,4. Cuando se unen a la AQP4 en el pie del astrocito se activa el complemento y se inicia el depósito de complejos de ataque a la membrana4. Las citocinas (IL 17, IL 8 y factor estimulante de colonias de granulocitos, entre otras) reclutan neutrófilos y eosinófilos en los espacios perivasculares para su degranulación, causando la muerte de los astrocitos4. Asimismo, esta pérdida conduce a la muerte de los oligodendrocitos, lo que provoca degeneración axonal y muerte neuronal4. Por último, existe un infiltrado de macrófagos (y posiblemente, células de la microglía) que fagocitan la mielina y los restos celulares4. Luego, con las lesiones maduras existe necrosis tisular completa e infiltrados extensos por macrófagos y astrocitos reactivos confinados alrededor AQP44. En un estudio12 que analizó lesiones desmielinizantes supratentoriales de 13 casos de NMO (3 biopsias de cerebro y 10 autopsias) vs EM (datos bibliográficos) por inmunohistoquímica se encontró que la NMO puede mostrar pérdida de AQP4 y astrocitos, depósitos de complemento dentro de los macrófagos y apoptosis de oligodendrocitos asociados con una pérdida selectiva de glucoproteína asociada a la mielina, datos que se asemejan a los patrones de lesión tipo ii y iii de la EM. Estas lesiones ocurren simultáneamente solo en un subconjunto de lesiones activas de NMO, y apoyan el concepto de heterogeneidad interindividual como en la EM.
La NMO se presenta clínicamente con ataques de NO (pérdida de la agudeza visual, dolor a la movilización ocular y discromatopsia) generalmente unilateral y LETM (tetra o paraplejía, nivel sensitivo y alteración de esfínteres). Cuando existe asociación de ambas por un lapso menor a 30 días el curso es llamado monofásico (10-40%). Sin embargo, frecuentemente se presenta en forma de brotes y remisiones (polifásica), completando el cuadro en el 60% de los casos al año y en el 90% a los 3 años8,9. El concepto tradicional de RM de cerebro «normal» (excluyendo los nervios ópticos) en la NMO ha cambiado desde que Pittock et al. encontraron que el 60% de los pacientes tenían lesiones inespecíficas asintomáticas en el SNC y el 10% tenían lesiones «atípicas» que involucraban el tálamo o el hipotálamo (diencéfalo), tronco o cerebelo correspondientes a regiones con alta expresión de AQP41,13. Asimismo, el 10% de los pacientes cumplían criterios por imágenes de EM1,9,13. Es interesante comentar 3 artículos recientes que describen la ausencia de desmielinización cortical en la NMO, dato que la diferencia de la EM. Popescu et al.14 en un estudio neuropatológico (serie de casos) que analizó por inmunohistoquímica autopsias de 19 pacientes con diagnóstico clínico y/o confirmación patológica de NMOSD en forma de corte transversal, visualizaron ausencia de desmielinización cortical cerebral y cerebelosa con conservación de la distribución de la AQP4. Este trabajo concluye que estos datos proporcionan una explicación posible para la ausencia de un episodio progresivo en la NMO. En otro estudio15 se evaluó in vivo la presencia de lesiones corticales (LC) en secuencias de inversión-recuperación y espesor cortical por la aplicación Freesurfer en T1 (volumetría por eco) sobre 90 pacientes (NMO: 30, EMRR: 30 y controles normales: 30), no encontrándose LC en controles normales ni en pacientes con NMO (leve adelgazamiento en circunvoluciones poscentral, precentral y calcarina, así como en el tálamo cuando se comparó NMO con controles normales). No fue así en pacientes con EM, donde 20/30 (66,7%) presentaron LC. Por lo tanto, el análisis de RM de la corteza es una herramienta fundamental, especialmente en los casos de diagnóstico indeterminado. Estos hallazgos fueron corroborados en otro estudio16 que comparó NMOSD (n=10) vs EM (n=18) en RM de 7 tesla bajo el protocolo de obtención de imágenes ponderadas en T2 (fast low angle shot) y secuencias de doble inversión-recuperación, donde se encontró que las lesiones en la sustancia blanca de la EM eran perivenulares (92%) en comparación a las de NMOSD (35%) que eran «paravenulares», con ausencia de enfermedad cortical en sustancia gris (NMOSD: 0 vs EM: 7/18).
Durante los ataques de mielitis con IgG-NMO positivo, la RM evidencia lesiones lineales que afectan 3 o más segmentos medulares (LETM), generalmente cérvico-dorsales, centromedulares y con realce del contraste17. Es importante tener en cuenta en qué momento se ha realizado dicha RM en relación con el ataque, porque cuanto más tarde se realice el estudio la lesión podrá dividirse (lesiones «parcheadas») en varios segmentos más cortos y no contiguos, como probablemente ocurrió en nuestra paciente18. Con respecto al LCR se puede encontrar pleocitosis prominente en el 25% de los pacientes, con predominio de células polimorfonucleares o monomorfonucleares, así como también bandas oligoclonales entre el 15-30% de los pacientes con NMO, como se presentó en nuestro caso1,8.
Los avances en la inmunopatogenia y en las imágenes por RM llevaron a revisar los criterios diagnósticos. Wingerchuck et al. plantearon que para definir a la NMO se necesita LETM más NO y al menos 2 de los siguientes criterios: RM de encéfalo inicial normal o que no cumpla con los criterios de Barkhof / Tintoré para EM, RM de médula con afección de al menos 3 segmentos medulares y/o IgG-NMO positivo en suero2. Es importante aclarar y destacar que si bien la IgG-NMO es de gran ayuda para el diagnóstico, no es fundamental porque aun sin este marcador específico en suero se puede llegar a definir la NMO. El descubrimiento de los anticuerpos IgG-NMO no solo ayudó a entender y caracterizar mejor la enfermedad, sino también a definir el espectro clínico asociado a la presencia de estos anticuerpos, con implicaciones pronósticas y terapéuticas. Ante un primer ataque de LETM y/o NO con IgG-NMO positiva existe un riesgo de recurrencia elevado de conversión a NMO definitiva. En un estudio prospectivo de 29 pacientes con un primer evento de LETM con IgG-NMO positivo se encontró que existe un 56% de riesgo de recurrencia de LETM o NO (conversión a NMO) durante el seguimiento a 12 meses6. Con respecto a NO el 20% de los pacientes con NO recurrente son positivos para IgG-NMO, y estos pacientes tienen un peor score visual y un 50% de riesgo a desarrollar mielitis transversa19. Además, este anticuerpo parece ser un marcador altamente específico para OSMS asiática, dado que un estudio de cohorte japonés encontró que el 58% fueron seropositivos en comparación con el 0% de las EM típicas o convencionales3,20. En resumen, la positividad para IgG-NMO es de aproximadamente un 50% en pacientes con recidiva de LETM y de un 25% en pacientes con NO simultánea o recurrente con RM de encéfalo negativa3,6,19. Cabe destacar que el hipo y las náuseas persistentes e intratables se pueden presentar en el 17-43% y el compromiso respiratorio puede generar paro respiratorio y muerte por extensión al tronco del encéfalo en un tercio de los pacientes2,21. Dentro del espectro de la NMO se encuentra la asociación con enfermedades autoinmunes órgano-específicas (miastenia gravis y tiroiditis autoinmune) y no órgano-específicas, como el lupus eritematoso sistémico (LES) y síndrome de Sjogren (SS)22. Asimismo, los anticuerpos que se encontraron con mayor frecuencia en un estudio que evaluó NMO y LETM fueron el antinuclear (43,8%) y el anti-RO/anti-LA (15,7%)7,22. En este estudio se observó una coexistencia de NMO y enfermedad autoinmune en el 28% de los pacientes. Algunos autores consideran que si el LES o el SS es clínicamente evidente o se encuentra una asociación con autoanticuerpos positivos, existiría una coexistencia de síntomas y signos de NMO en la cual el proceso neurológico se basaría en una complicación vasculítica de la enfermedad sistémica1,3,4,22. Sin embargo, estudios recientes demostraron que la seropositividad de IgG-NMO no aparece en individuos con LES o SS clínicamente definido, y estos carecen de síntomas clínicos del espectro NMO7,22. Además, la IgG-NMO se detecta con mayor frecuencia en los pacientes con síntomas de NMO que tienen evidencia clínica o serológica de LES o SS que en aquellos que no la tienen. Por lo tanto, la asociación de NMO seropositiva con enfermedades autoinmunes (LES o SS) representa una coexistencia de 2 enfermedades autoinmunes más que una complicación vasculítica secundaria de la enfermedad sistémica1,3,4,22.
Con respecto a la terapéutica, ante un brote o ataque de NO o LETM debemos considerar el tratamiento en el agudo y el preventivo en las nuevas recaídas. Es importante aclarar que no existen estudios clínicos con alto nivel de evidencia; por lo tanto, estos tratamientos se basan en recomendaciones de expertos. Con respecto al primero, la medida terapéutica de primera línea es la metilprednisolona 1 g/d endovenosa (EV) por 3 a 5 días consecutivos con buena respuesta en más del 80% de los pacientes8,12,23. Típicamente los ataques de mielitis se tratan por 5 días y los de NO durante 3 días consecutivos seguidos de un mantenimiento oral18,24. Cuando estos ataques son severos y no responden a los corticoesteroides la evidencia marca que la plasmaféresis sola o en combinación con prednisona de forma precoz es segura, eficaz y se asocia a mejor pronóstico funcional en el 50-60% de los pacientes25–27. La inmunoglobulina intravenosa (IgIV) es utilizada en casos refractarios a esteroides. Sin embargo, esta terapia no fue apoyada por un ensayo pequeño de 76 pacientes que combinaba IgIV y metilprednisolona. La evidencia actual no apoya el papel principal de la IgIV en el tratamiento de los ataques agudos de NMO, aunque sería razonable tratar a pacientes refractarios o que presenten contraindicaciones a otras terapias para el tratamiento agudo27. El tratamiento preventivo está indicado en pacientes con enfermedad recidivante o que tengan un alto riesgo de desarrollar enfermedad recurrente, como fue descrito anteriormente. La combinación de azatioprina (AZA) más prednisona es lo más utilizado. La dosis de inducción para la prednisona es 1mg/kg/d durante 4 a 6 meses y luego se recomienda una reducción de no más de 5mg/semana y posteriormente reducir 5mg cada 2 semanas hasta dosis de 5-20mg/d o en días alternos (se asoció a reducción de la tasa de recaídas en la NMO) y discontinuarla si es posible28. La AZA a dosis de 2-3mg/kg/d es el inmunosupresor más utilizado para prevenir los ataques de NMO y evitar los efectos adversos de los corticoides a largo plazo, y se asocia a una mejor funcionalidad neurológica en la Expanded Disability Status Scale (EDSS)29. Estos datos son avalados por un estudio prospectivo reciente30 sobre 99 pacientes con NMOSD (NMO: 86, NMOSD seropositivos: 13) con seguimiento a un año (solo 70 pacientes tuvieron seguimiento mayor de un año), donde el 37% no presentó recaídas (rango: 12-154 meses; mediana: 24), en 22 pacientes se mantuvo estable o mejoró su EDSS y en 54 pacientes mejoró su score visual. Se suspendió el tratamiento en 38 pacientes (falta de eficacia y efectos adversos). Por lo tanto, la AZA se debería iniciar tempranamente en el NMOSD dado que es efectiva, bien tolerada, mejora el EDSS y el score visual. El micofenolato en dosis de 1-3mg/kg/d es utilizado ante efectos secundarios o contraindicación para la AZA (toxicidad inducida por déficit de la tiopurina metiltransferasa)31. El rituximab (anticuerpo monoclonal anti-CD20) ha recibido un interés creciente dada su selectividad para la depleción de células B. Si bien algunos autores lo postulan como primera línea de tratamiento, la tendencia indica que es usado ante enfermedad severa con pobre respuesta a otros inmunosupresores32,33. Existen 2 opciones de administración. El protocolo utilizado para el tratamiento de linfoma, con administración EV de 375mg/m2 por semana durante 4 semanas y el protocolo para artritis reumatoide, con infusión EV de 1.000mg la primera semana e igual dosis repetida a las 2 semanas. Para ambas opciones la reinfusión se realiza cada 6-12 meses. Aunque la duración óptima del tratamiento se desconoce, según las publicaciones es de 2 años8.
En conclusión, el descubrimiento de la presencia de los anticuerpos IgG-NMO no solo contribuyó a la mejor caracterización de la NMO y su distinción con la EM, sino también a ampliar el espectro clínico de las enfermedades asociadas. Esto es de gran valor en la práctica clínica, fundamentalmente ante el primer evento de NO o LETM, debido a que la seropositividad cambia la conducta médica. En estos casos la terapia preventiva con fármacos inmunosupresores debería ser instituida tan rápido como sea posible, dado que la discapacidad se relaciona con el efecto residual de los ataques.
Conflicto de interesesLos autores declaran que no existe ningún conflicto de intereses.
