




La polineuropatia amiloidótica familiar por transtiretina (PAF-TTR), enfermedad autosómica dominante. generalmente se manifiesta con miocardiopatía y polineuropatía; sin embargo, síntomas gastrointestinales (GI) son frecuentes. Reportamos paciente con AF-TTR y síntomas GI de inicio.
Caso clínicoHombre de 67 años, consulta por dolor neuropático urente. Antecedentes: diarrea alternando con constipación y pérdida de peso, diagnóstico intestino irritable. Examen físico: alteración pruebas sensibilidad frio-calor. Estudio de conducción nerviosa polineuropatía axonal sensoriomotora. Análisis gen transtiretina variante heterocigota p.Val50Met, diagnóstico PAF-TTR.
ConclusionesLa no especificidad de síntomas GI al inicio del cuadro son un desafío en la sospecha diagnóstica de esta entidad.
Familial transthyretin amyloidotic polyneuropathy (FAP-TTR), an autosomal dominant disease. It generally manifests with cardiomyopathy and polyneuropathy, however, gastrointestinal (GI) symptoms are common. We report a patient with AF-TTR and initial GI symptoms.
Clinical caseA 67-year-old man presented with burning neuropathic pain. History, diarrhea alternating with constipation and weight loss, diagnosis of irritable bowel. Physical examination alteration of hot-cold sensitivity tests. Sensorimotor axonal polyneuropathy nerve conduction study. Transthyretin gene analysis heterozygous variant p.Val50Met, PAF-TTR diagnosis.
ConclusionsThe non-specificity of GI symptoms at the onset of the condition is a challenge in the diagnostic suspicion of this entity.
La polineuropatía amiloidótica familiar por transtirretina (PAF-TTR) es una enfermedad multisistémica de base genética descrita en 1952 por Corinho Andrade en pacientes del norte de Portugal, donde se conocía popularmente como mal dos peshinos. Originalmente considerada como una condición endémica en algunas áreas de Portugal, Japón y Suecia, la enfermedad se manifiesta a nivel mundial con una menor frecuencia.
Se trasmite generalmente en forma autosómica dominante, aunque existen mutaciones de novo y casos esporádicos. Es causada por una variante patogénica en el gen TTR, que se encuentra en cromosoma 18q12.1 y comprende 4 exones y 5 intrones que codifican para la proteína transtirretina. Hasta la fecha se describen más de 150 variantes patogénicas de TTR que causan la formación de amiloide; sin embargo, la sustitución de valina por metionina en la posición30 es la variante más común (p.Val50Met)1-4.
La transtirretina es la proteína precursora del amiloide y actúa normalmente como transportador de la tiroxina y de la vitaminaA; esta se sintetiza de forma mayoritaria en el hígado (95%), y en un pequeño porcentaje en la retina y en los plexos coroideos. Tanto en la sangre como en el líquido cefalorraquídeo (LCR) circula en forma de un complejo tetramérico. La transtirretina mutada lleva a que se desestabilice el tetrámero, dando lugar a monómeros que se pliegan anormalmente y precipitan de forma parcheada en diferentes tejidos, principalmente en el sistema nervioso periférico y en el corazón3.
La PAF-TTR se manifiesta con una variedad de presentaciones clínicas, de las cuales la miocardiopatía y la polineuropatía son las más comunes2. Debido a la heterogeneidad del fenotipo, el diagnóstico puede retrasarse de 4 a 5años, especialmente en áreas no endémicas, y el diagnóstico erróneo es una carga frecuente para tales pacientes5.
Los pacientes suelen presentar síntomas predominantemente neurológicos y/o cardíacos; sin embargo, los síntomas gastrointestinales (GI) no específicos debido a afectación autonómica y síntomas constitucionales son comunes. Según la edad de inicio de los síntomas la enfermedad puede clasificarse como de inicio temprano (<50años) o de inicio tardío (≥50años). Sin tratamiento, la esperanza de vida promedio es de 7 a 11años desde el inicio de los síntomas1-6. Con la aprobación de terapias efectivas que pueden estabilizar la enfermedad y reducir la mortalidad en PAF-TTR, es de extrema importancia la identificación temprana de los pacientes afectados4.
Nuestro objetivo es presentar el caso de un paciente con PAF-TTR que inicialmente se presentó con síntomas predominantemente gastrointestinales y constitucionales, siendo los síntomas neurológicos leves al inicio del cuadro, lo que llevó a retraso significativo en el diagnóstico.
Caso clínicoUn hombre de 67 años, que consulta a servicio de neurología por presentar dolor de tipo urente en miembros a predominio de ambos pies, al inicio de predominancia nocturno para posteriormente ser continuo durante el día. Se habían realizado electromiografías normales, se encontraba en estudio sin etiología demostrada y presentaba una evolución de aproximadamente dos años, siendo interpretado como neuropatía de fibra fina idiopática. No presenta antecedentes familiares de dolor neuropático y refiere de aproximadamente cuatro años de evolución episodios de diarrea con 8 a 10 deposiciones diarias con dolor abdominal leve durante la defecación, alternando con periodos de constipación asociados a distensión abdominal, que fue interpretado como posible intestino irritable mixto por presentar colonoscopias y TAC de abdomen normales. Refiere además pérdida de aproximadamente 10kg de peso.
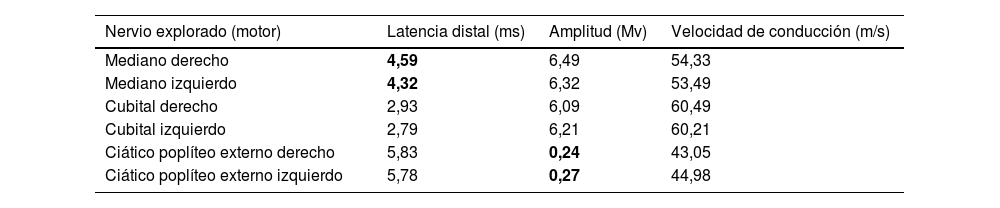
Al examen clínico se demostró alteración en las pruebas de sensibilidad frío-calor en distribución en botas, sin debilidad muscular y sin alteraciones de la marcha. Su estudio de neuropatía incluyó vitaminaB12, hemoglobinaA1c, función tiroidea, función renal, hepática, cadenas livianas en sangre y la electroforesis de proteínas en suero/orina de 24horas con resultado normal. Se solicita estudio de conducción nerviosa, demostrándose una polineuropatía axonal sensoriomotora en ambos miembros inferiores con signos de compromiso de ambos nervios medianos compatible con síndrome del túnel del carpo de grado leve bilateral (tabla 1).
Resultado de la electromiografía digital de los cuatro miembros con velocidad de conducción
| Nervio explorado (motor) | Latencia distal (ms) | Amplitud (Mv) | Velocidad de conducción (m/s) |
|---|---|---|---|
| Mediano derecho | 4,59 | 6,49 | 54,33 |
| Mediano izquierdo | 4,32 | 6,32 | 53,49 |
| Cubital derecho | 2,93 | 6,09 | 60,49 |
| Cubital izquierdo | 2,79 | 6,21 | 60,21 |
| Ciático poplíteo externo derecho | 5,83 | 0,24 | 43,05 |
| Ciático poplíteo externo izquierdo | 5,78 | 0,27 | 44,98 |
Se plantea el diagnóstico PAF-TTR por presentar signos de dolor neuropático, polineuropatía axonal y compromiso de fibras autonómicas, por lo que se evalúa el gen de TTR mediante ampliación y secuenciación de exones2, 3 y4 del gen, demostrándose una variante heterocigota p.Val50Met c.148G>A, lo que confirma el diagnostico.
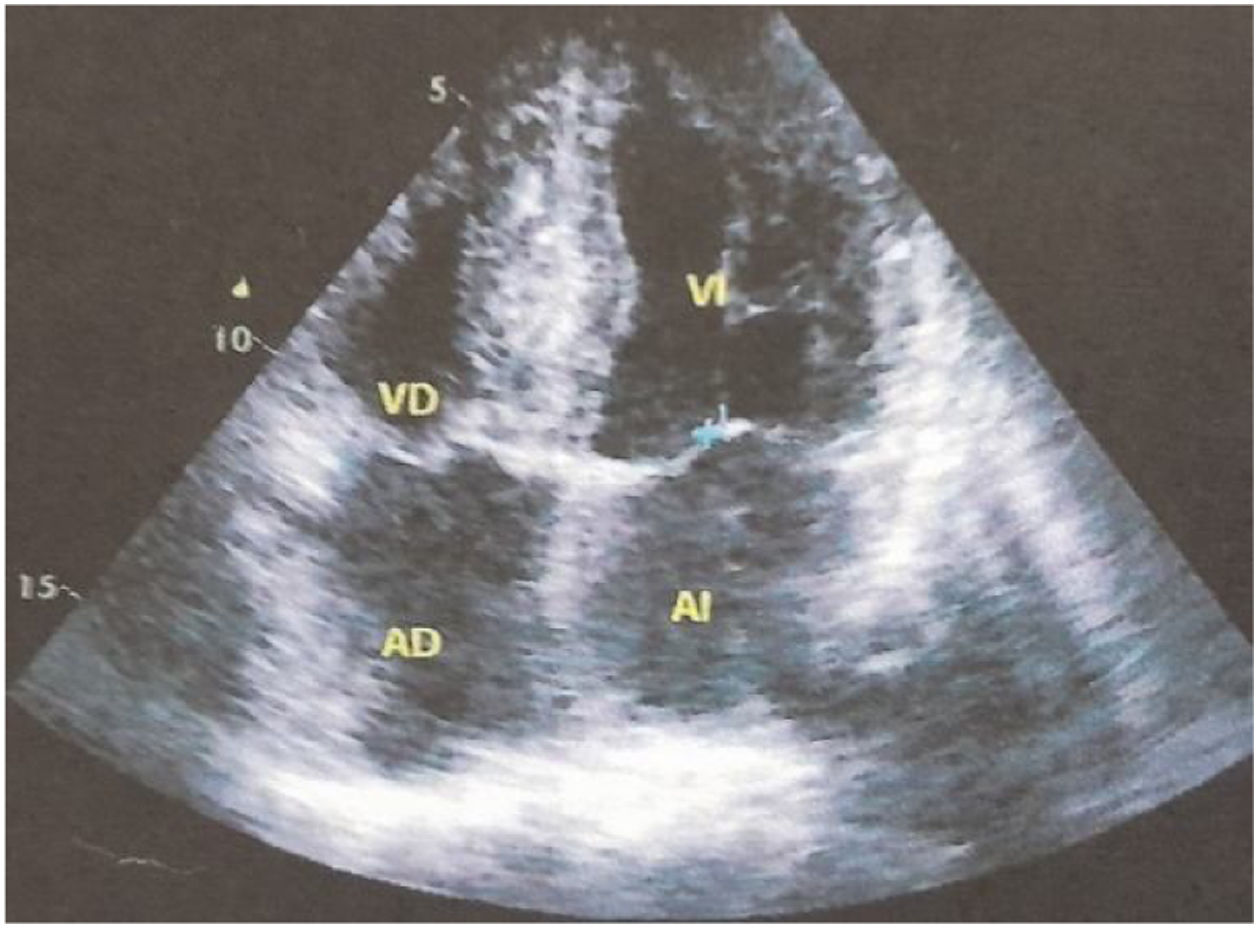
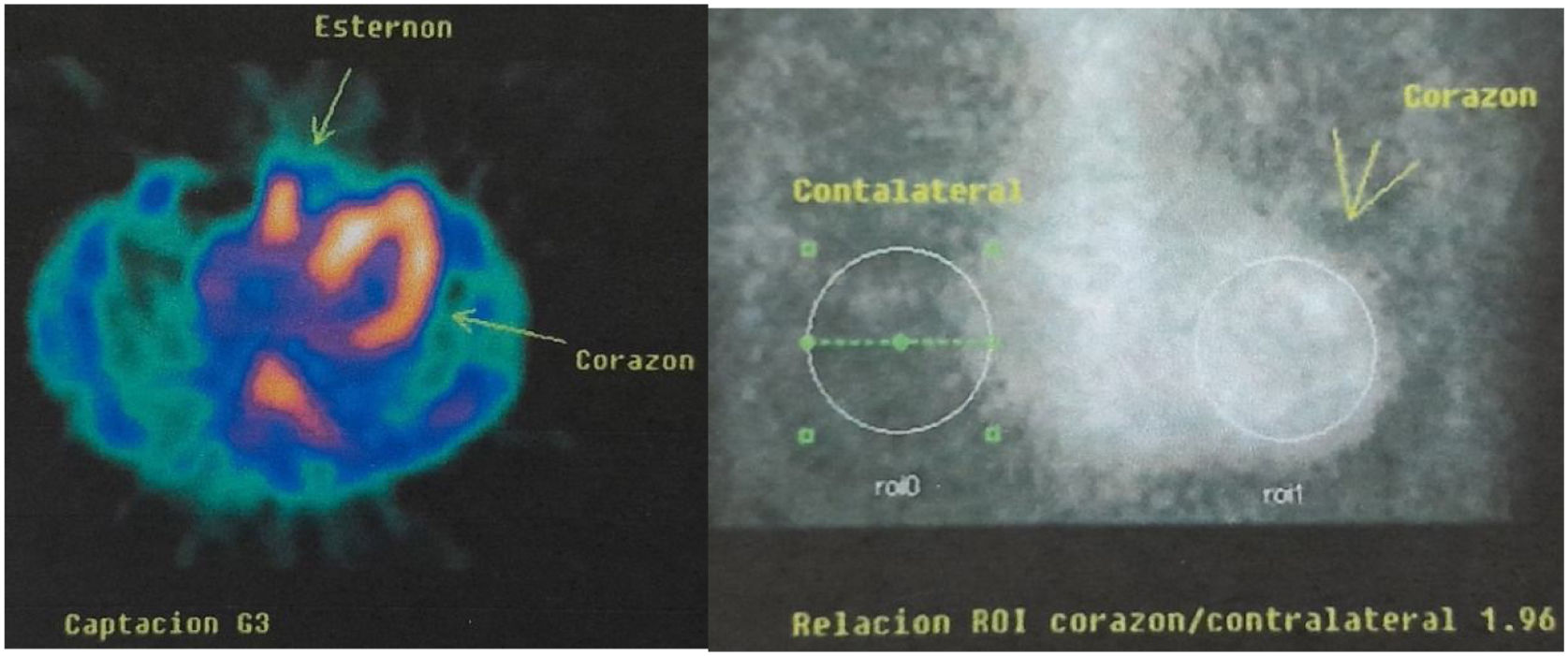
Para evaluar el posible compromiso cardiológico de la enfermedad se solicita ECG Holter de 24horas; ritmo sinusal, intervalo PR promedio 290ms (bloqueo auriculoventricular 1 grado), QRS 140ms, frecuencia cardiaca promedio 65lat/min. El ecocardiograma transtorácico demostró función ventricular normal: fracción de eyección del 72%, aumento severo del espesor parietal del tabique interventricular basal y medial con un diámetro diastólico de 21mm, resto de paredes ventriculares con espesor parietal 11mm, dilatación leve aurícula izquierda (21cm2) y de aorta ascendente (fig. 1). Se confirma el compromiso cardiaco mediante Spect cardiaco con tecnecio-99m-pirofosfato, que arroja resultados compatibles con el depósito de amiloidosis TTR cardíaco, relación nivel de captación del radiotrazador del área cardiaca en relación con el área torácica contralateral de 1,96 (valor normal, menor de 1,5) (fig. 2).
Discusión

Los síntomas GI pueden estar presentes incluso antes del inicio de la polineuropatía periférica en los casos de PAF-TTR, principalmente en aquellas zonas no endémicas de la enfermedad. La diarrea puede ser el primer síntoma, siendo al inicio posprandial y en etapas posteriores estar acompaña de incontinencia fecal o episodios alternantes de diarrea con constipación. Estos síntomas son inespecíficos y es difícil diferenciarlos de otras patologías gastrointestinales mucho más frecuentes en su incidencia que pueden producirlos. En este caso, el cuadro se interpretó como secundario al diagnóstico de intestino irritable mixto, ya que cumplía con los criterios de RomaIV, diagnósticos de esta entidad. Es para destacar que la pérdida de peso es una bandera roja en el posible diagnóstico de intestino irritable, por lo que se deberían buscar etiologías alternativas. La pérdida de peso es una manifestación frecuente en PAF-TTR, siendo hasta más frecuente que la diarrea y de causa multifactorial (desnutrición, saciedad precoz por gastroparesia), siendo observado incluso antes del inicio de otros síntomas. La patogenia de las manifestaciones GI no están del todo aclaradas; sin embargo, se sugieren alteraciones de la motilidad debidas a la denervación de los nervios autónomos7,8, aumentan su incidencia con la duración de la enfermedad y se asocian con una disminución de la calidad de vida9,10.
El compromiso autonómico puede afectar el sistema cardiocirculatorio desarrollando hipotensión ortostática, que puede ser asintomática o manifestarse con mareos al ponerse de pie, fatiga o visión borrosa. La gastroparesia y los vómitos posprandiales causan deshidratación y aumentan el riesgo de hipotensión ortostática y de pérdida de peso progresivo secundaria a saciedad temprana10. Puede haber disuria y episodios de retención de orina o urgencia miccional. La disfunción sexual puede aparecer en forma temprana hasta antes de los síntomas sensitivos, principalmente en el varón. A la evaluación oftalmológica pueden demostrarse pupilas festoneadas e irregulares secundarias a depósito de amiloide en los nervios ciliares1,8,9.
En las regiones endémicas la enfermedad se manifiesta a partir de los 30 a los 40años, siendo la forma más frecuente de presentación la de una neuropatía sensitiva con sensación de entumecimiento de los pies y episodios de dolor neuropático como principal manifestación clínica secundario a la afectación inicial de fibras nerviosas finas mielínicas y amielínicas encargadas de la transmisión del dolor y de la temperatura. El dolor se caracteriza por ser de tipo quemante, asociado a alodinia, y puede tener un ritmo cicardiano, con empeoramiento en las últimas horas del día. En esta etapa de la enfermedad se pueden demostrar al examen clínico alteraciones en la evaluación de la sensibilidad térmica y dolorosa, con preservación de la sensibilidad al tacto y la propiocepción. Los síntomas progresan de manera ascendente hasta comprometer las piernas, los muslos y posteriormente los miembros superiores. A medida que el depósito de material amiloide compromete axones largos se desarrolla una afectación de las funciones motoras con compromiso progresivo de la marcha. Meses a años posteriores el déficit motor progresa con la evolución natural de la enfermedad, dificultando el poder caminar sin ayuda. La pérdida de la sensación al dolor asociada a una fuerza muscular normal o subnormal puede llevar a que los pacientes desarrollen ante el trauma úlceras plantares y osteoartropatía del pie (artropatía de Charcot)7-8. En zonas no endémicas la enfermedad puede aparecer de forma tardía (por encima de los 50años) y cursar con una polineuropatía motora o sensitiva pura, sin manifestaciones autonómicas, o presentarse como mononeuropatías que progresan hacia una mononeuritis múltiple confluyente y que acaban formando un patrón polineuropático1-3.
El compromiso inicial de fibras finas explica el resultado normal de las primeras electromiografías realizadas en el paciente que describimos, y posteriormente el compromiso es típicamente axonal. No es raro que un moderado enlentecimiento en la velocidad de conducción nerviosa se sobreinterprete y valore como una forma de polineuropatía desmielinizante crónica (CIDP), quizá por el afán de ofrecer una posibilidad terapéutica. La CIDP fue el diagnóstico erróneo más frecuente de PAF-TTR, ya que las características desmielinizantes en el estudio de la conducción nerviosa y un leve aumento de las proteínas del LCR resultaron ser un factor relevante que condujo al diagnóstico erróneo de la enfermedad. Sin embargo, no es entendido completamente si los cambios de velocidad de conducción con su reducción representan una verdadera desmielinización o son principalmente secundarios a pérdida de conducción rápida de las fibras de gran diámetro11.
En el 80% de PAF-TTR se manifiestan síntomas cardíacos, pudiendo presentarse con insuficiencia cardíaca restrictiva o arritmia. El fenotipo clásico es insuficiencia cardíaca con fracción de eyección preservada, y se han detectado bloqueos de la conducción auriculoventricular con episodios sincopales que han requerido marcapasos, e incluso muerte súbita. El compromiso cardiaco parece ser más común en mutaciones Val50Met en hombres de inicio tardío. Un grosor del tabique interventricular >12mm en ausencia de enfermedad hipertensiva o patología de la válvula aórtica es el criterio diagnóstico mediante ecocardiografía que corrobora la participación cardiaca en pacientes con PAF12.
A pesar de que es una enfermedad rara y de base genética, se debe mantener un alto índice de sospecha en pacientes adultos con síntomas y síntomas compatibles, a pesar de no tener historia familiar de la enfermedad. La presencia de antecedentes familiares ayuda en la sospecha diagnóstica frecuentemente en los casos endémicos; sin embargo, los casos de inicio tardío suelen ser esporádicos, como es el caso del paciente presentado. El hecho de que la penetrancia sea incompleta y que la edad de inicio pueda ser muy tardía, de modo que algunos portadores de la mutación no lleguen a desarrollar los síntomas porque fallezcan antes, puede dar lugar a la detección de casos aparentemente esporádicos. También puede ocurrir que los antecedentes familiares no se reporten por desconocimiento. La medicina nuclear mediante el uso de spect cardiaco con pirofosfato de tecnecio es una herramienta relativamente novedosa que tiene una alta sensibilidad para evaluar el compromiso cardiaco en PAF-TTR. Se ha demostrado que la captación miocárdica de fosfonatos tiene una excelente correlación con la biopsia cardíaca, con una especificidad y un valor predictivo positivo del 100% para el diagnóstico de amiloidosis TTR. Se puede evaluar el grado de captación cardíaca mediante dos métodos. Uno es visual o semicuantitativo (de acuerdo a la escala visual de Perugini), donde se compara la captación miocárdica con la observada a nivel de estructuras óseas, clásicamente costillas y esternón, y se asigna un grado de captación, a saber: grado 0=ausencia de captación a nivel miocárdico; grado 1=captación leve, menor que el tejido óseo; grado 2=captación moderada, similar al tejido óseo; grado 3=captación significativa, mayor que el tejido óseo. El otro método es cuantitativo y consiste en determinar un índice de captación que relaciona un área cardíaca (H) respecto de un área similar en el hemitórax contralateral (CL). La relación H/CL >1,5 es altamente sugestiva para amiloidosis TTR5. Esta técnica puede demostrar, por su alta sensibilidad y especificidad, el compromiso cardiaco en PAF-TTR a pesar de que la ecocardiografía o la RMI cardiaca sean normales. De hecho, no son raros los casos diagnosticados de forma incidental a raíz de una gammagrafía realizada por indicaciones oncológicas o reumatológicas13-14.
Las pruebas genéticas se han convertido en la prueba diagnóstica de elección para pacientes que presentan neuropatía, dado la baja sensibilidad de muchos sitios de biopsia de tejido. El test diagnóstico de elección es la secuenciación completa del gen de la TTR, dado que es un gen pequeño y, con el desarrollo actual de las técnicas de diagnóstico genético, es un estudio barato, sencillo, rápido y con una elevada sensibilidad y especificidad. Dado que, especialmente en zonas no endémicas, la enfermedad puede presentarse como formas atípicas de inicio tardío y sin antecedentes familiares conocidos, creemos que el estudio genético de TTR debe incluirse en el protocolo diagnóstico de cualquier polineuropatía. En caso de confirmarse la enfermedad, se debe realizar un estudio genético de los familiares de riesgo y aportar el consejo genético necesario1-9.
Este caso pone de relieve múltiples desafíos que pueden retrasar el diagnóstico de PAF-TTR. Los síntomas iniciales a menudo no son específicos de compromiso gastrointestinal, los síntomas neuropáticas son leves y los antecedentes familiares pueden estar ausentes o ser engañosos. No obstante, la identificación temprana sigue siendo importante, ya que las terapias recientemente aprobadas (tafamidis, inotersén, patisirán) están diseñadas para reducir el depósito adicional de amiloide pero no abordan el efecto del amiloide ya depositado4.
ConclusionesDescribimos un caso de PAF-TTR asociado a mutación Val50Met, que inicialmente se presenta con síntomas GI prominentes y posteriormente desarrolla signos neuropáticos. El diagnóstico fue realizado a los cuatro años desde el momento de la presentación de los síntomas, lo que estaría dentro del rango de 1 a 10años de retraso en el diagnóstico descripto en la literatura, pero es de extrema importancia tener en cuenta esta entidad en aquellos pacientes con síntomas GI inespecíficos sin etiología determinada para poder realizar un diagnóstico precoz y evitar las complicaciones propias de la enfermedad, ya que las terapias no son efectivas sobre la transtirretina ya depositada11-14.
Sin tratamiento, PAF-TTR es una enfermedad mortal, y este caso enfatiza la importancia de tener un alto índice de sospecha al considerar su diagnóstico. Los pacientes que presentan combinación de neuropatía asociado con uno o más de los siguientes ítems: 1)antecedentes familiares; 2)signos de disfunción autonómica temprana, signos de miocardiopatía, pérdida de peso sin explicación, y 3)síndrome del túnel carpiano u opacidad del vítreo, deben ser estudiados15.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
