





Las enfermedades mitocondriales representan un grupo de trastornos multisistémicos, que frecuentemente incluyen disfunción del sistema nervioso central y periférico entre sus manifestaciones, causados por diversas mutaciones localizadas tanto en el genoma mitocondrial como en el nuclear. La amplia variabilidad observada en su expresión clínica y la amplitud etiopatogénica complica su diagnóstico molecular.
ObjetivosPresentar un algoritmo diagnóstico que permita seleccionar la secuencia de estudios moleculares que pueden realizarse en la individualización diagnóstica molecular de los trastornos mitocondriales con manifestaciones en el sistema nervioso.
DesarrolloLa aproximación diagnóstica de los trastornos mitocondriales puede simplificarse si se contestan en forma secuencial 3 preguntas. ¿Cuál es la probabilidad de que mi paciente tenga un trastorno mitocondrial? Utilizando criterios diagnósticos e índices de sospecha de enfermedad mitocondrial puede estimarse la probabilidad de que la presentación clínica y los resultados de algunos estudios complementarios correspondan a un trastorno mitocondrial. Si tengo esta sospecha, ¿tiene mi paciente una presentación sindromática clásica? Si mi paciente presenta hallazgos característicos de síndromes mitocondriales clásicos, el orden de estudios moleculares por ser solicitados puede considerarse relativamente establecido. Si no tiene presentación sindromática, ¿es un defecto aislado de la cadena respiratoria? En esta situación, los resultados de pruebas bioquímicas de funcionamiento de los distintos complejos de la cadena respiratoria dirigirá la sistematología de pruebas moleculares solicitadas.
ConclusionesLa respuesta a estas 3 preguntas permitirá sistematizar y dirigir el orden y extensión de las pruebas moleculares necesarias para la individualización diagnóstica molecular de los trastornos mitocondriales.
Mitochondrial disorders are a group of multisystemic diseases that frequently show central and peripheral nervous system compromise, caused by several mitochondrial and nuclear genome mutations. Diverse and heterogeneous clinical presentations along a broad group of molecular causes make complex its diagnosis.
ObjectivesTo present a diagnostic algorithm useful in the decision of which molecular test could be ordered in the molecular diagnosis of mitochondrial disorders manifesting in the nervous system.
Main ConceptsMitochondrial disorders diagnostic's approach could be simplified if three questions are sequentially answered. Which is the probability that my patient have a mitochondrial disorder? Using diagnostic criteria and suspicion indexes, a probability of mitochondrial pathology could be estimated from clinical symptoms and complementary tests results. If I think my patient has a mitochondrial disorder, does he show a classical presentation corresponding to a well-described syndrome? The group of molecular tests to be ordered in patients showing classical presentations could be considered well established. If not, does he suffer from an isolated defect of one subunit of the respiratory chain complex? The results obtained from biochemist tests assessing the function of each subunit of the respiratory complex must direct the molecular tests to be ordered.
ConclusionsThe answers to these three questions let systematize and direct the molecular diagnostic approach of mitochondrial disorders.
Cómo ha sido la evolución de los organismos eucariotas en general y de sus mitocondrias en particular es una pregunta importante de la biología evolutiva que no posee una respuesta definitiva todavía1. Tradicionalmente se acepta que las mitocondrias son un resabio evolutivo de una bacteria endosimbiótica perteneciente al grupo de las alfaproteobacterias, ancestros de Escherichia y Neisseria, representando una huella de la transición procariota-eucariota en la historia de la vida2. Sin embargo, la presencia en nuestros genomas de múltiples genes originarios de distintas especies bacterianas permite cuestionar la validez absoluta de esta teoría monobacteriana3.
De todos modos, el presente de nuestras células eucariotas muestra que coexisten 2 genomas, el nuclear y el mitocondrial4. Diversos trastornos del sistema nervioso central y periférico -pleomórficos en sus manifestaciones clínicas- están causados por mutaciones en el genoma mitocondrial5. El genoma mitocondrial está formado por 16.569 pares de bases que codifican distintos ARN de transferencia y ribosomales funcionantes en la transcripción de 13 proteínas constituyentes de la cadena respiratoria. Además de la presencia de un código genético independiente, la genética mitocondrial es particular por la transmisión exclusiva matrilínea y la presencia de mutaciones patogénicas en heteroplasmia (coexistencia en distintas proporciones de genomas mitocondriales con variantes en su secuencia) dependiente del tejido6. Precisamente las particularidades de la heteroplasmia, así como la relativa gran extensión nucleotídica por ser analizada, dificultan técnicamente el diagnóstico molecular de los trastornos mitocondriales7. Por otro lado, el número creciente de enfermedades del sistema nervioso consecuencia de una disfunción mitocondrial causadas por mutaciones en genes codificados en el núcleo complica aún más la aproximación diagnóstica en esta área8.
A continuación presentaré un enfoque diagnóstico secuencial mediante la respuesta a 3 preguntas que intentará servir como una recomendación práctica para el diagnóstico de los trastornos mitocondriales que tienen como manifestación síntomas del sistema nervioso.
¿Cuál es la probabilidad de que mi paciente tenga un trastorno mitocondrial?Como sucede con otros trastornos de la neurología, existen criterios diagnósticos o escalas de probabilidad diagnóstica que han sido desarrollados con el objetivo de establecer grados de certeza o posibilidad de la existencia de una disfunción mitocondrial como la etiología de la sintomatología experimentada por el paciente asistido. En la figura 1 se presenta una versión resumida de la escala de Wolf modificada9. Puede observarse que la suma de signos y síntomas indicativos de afectación muscular, del sistema nervioso central y multisistémica y los hallazgos en las pruebas bioquímicas e histopatológicas permite estratificar la probabilidad diagnóstica de una encefalomiopatía mitocondrial.

Criterios de probabilidad de disfunción mitocondrial a partir de datos clínicos y de estudios complementarios. Modificada de Wolf et al.9.
Entre los síntomas de afectación muscular, destacan la afectación oculomotora y la intolerancia al ejercicio; la afectación del sistema nervioso central frecuentemente incluye síntomas como ataxia, crisis epilépticas, mioclonías junto a pérdida de pautas madurativas y fenómenos stroke like; característicamente otros sistemas se ven afectados con sintomatología que indica disfunción cardíaca, pancreática, renal y auditiva10–13.
Algunos hallazgos en las pruebas bioquímicas permiten incrementar la sospecha de enfermedad mitocondrial: elevación de los niveles séricos y en LCR de ácido láctico, aumento en los niveles de alanina en la cromatografía de aminoácidos y aumento en la excreción de ácidos orgánicos como el etilmalónico en la orina14,15. Por otro lado, los aumentos en los niveles de ácido láctico en el sistema nervioso central también pueden observarse por medio de la espectroscopia por RM16.
Finalmente, la histopatología del tejido muscular constituye uno de los principales métodos complementarios para el enfoque diagnóstico de la enfermedad mitocondrial. Hallazgos característicos son las fibras rojo rasgadas y la disminución de la actividad oxidativa COX y SDH junto a la presencia de acumulaciones mitocondriales subsarcolemales y alteraciones en la morfología mitocondrial en la microscopia electrónica17.
La suma de los signos y síntomas junto a los hallazgos en los estudios complementarios permite obtener una puntuación total en la escala de Wolf modificada. Cuanto mayor es esta puntuación, mayor resulta ser la probabilidad de disfunción mitocondrial como etiología del trastorno asistido.
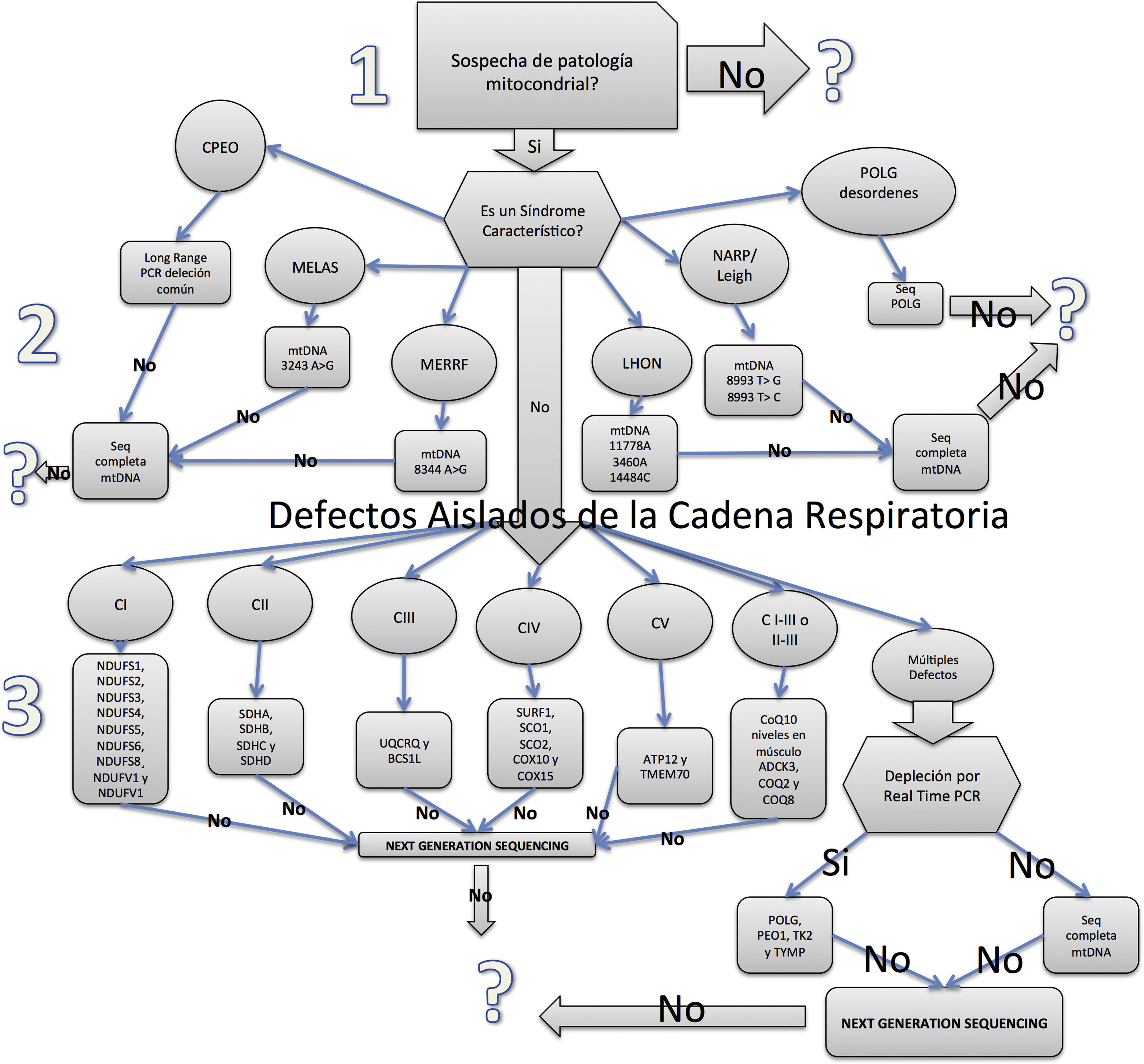
¿Tiene mi paciente una presentación sindromática clásica?Pese a la frecuente heterogeneidad sintomática y genética de la enfermedad mitocondrial, existen síndromes bien reconocidos que permiten seleccionar y dirigir la solicitud de pruebas moleculares diagnósticas de los trastornos mitocondriales.
MELASEste síndrome, originalmente descrito por Pavlakis et al.18, presenta como manifestación neurológica más frecuente la aparición de fenómenos stroke-like en los cuales sintomatología deficitaria focal se acompaña de lesiones hiperintensas, generalmente parietooccipitales, en las imágenes por RM sin un claro respeto de un territorio vascular y que evolutivamente no resultan persistentes19. Otras manifestaciones prevalentes en este síndrome son la hipoacusia neurosensorial, la diabetes, el deterioro cognitivo, la baja talla y la migraña20. En la mayoría de los pacientes que presenten este síndrome, una mutación en la posición 3243 del genoma mitocondrial correspondiente al gen codificante del ARN de transferencia para leucina en distintos niveles de heteroplasmia tisular es la causa del trastorno21. Los niveles de heteroplasmia suelen ser menores en los tejidos de alto recambio como el sanguíneo, y en consecuencia la búsqueda de esta mutación debe hacerse preferentemente en tejidos de menor recambio como el músculo o el epitelio vesical (sedimento de orina)22.
MERRFEste síndrome, originalmente descrito por Fukuhara et al.23, presenta como manifestación neurológica más frecuente el desarrollo de una epilepsia mioclónica progresiva junto a otras manifestaciones indicativas de disfunción mitocondrial: hipoacusia, miopatía, neuropatía y demencia24. Además, se ha descrito la presencia de lipomas axiales25 y síntomas psiquiátricos en pacientes afectados por este síndrome26. La mutación más frecuentemente causante de MERRF es la sustitución de una adenina por una guanina en la posición 8344 del genoma mitocondrial, codificante del ARN de transferencia para lisina27. Los niveles de heteroplasmia de esta mutación también muestran heterogeneidad tisular, aunque esta mutación particular puede detectarse en sangre con mayor sensibilidad que otras28.
Oftalmoplejía externa crónica progresiva y síndrome de Kearns- SayreLa oftalmoplejía externa crónica progresiva (CPEO) y el síndrome de Kearns-Sayre (SKS) característicamente muestran compromiso de la motilidad ocular externa: ptosis y oftalmoplejía10. Un comienzo antes de los 20 años de edad y la presencia de retinopatía pigmentaria junto a la variable aparición de trastornos en la conducción cardíaca, hiperproteinorraquia y ataxia, definen al SKS29. Además, los pacientes afectados por estos síndromes pueden mostrar otra sintomatología característica de disfunción mitocondrial, ya mencionadas en los párrafos precedentes. La mayoría de los sujetos afectados por CPEO y SKS presentan como causa una deleción de 4,9 kb en el genoma mitocondrial30. Esta deleción no puede ser determinada en muestras provenientes de sangre, requiriéndose tejido muscular para el diagnóstico. La mayoría de los sujetos afectados por estos síndromes y con esta alteración estructural en el genoma mitocondrial no presentan riesgo de recurrencia familiar31. Sin embargo, definitivamente existen formas familiares de CPEO. Cuando el análisis del familigrama muestra un patrón de herencia indicativo de afectación autosómica, dominante más frecuentemente y recesivo en menor medida, debe analizarse la secuencia de los genes PEO1, TK y POLG32. En cambio, un familigrama indicativo de herencia materna debe llevar al análisis de la secuencia completa del genoma mitocondrial33.
Neuropatía óptica hereditaria de LeberLa neuropatía óptica hereditaria de Leber (LHON) característicamente se presenta como una pérdida aguda-subaguda e indolora de la agudeza visual en sujetos jóvenes. Frecuentemente afecta a ambos nervios ópticos en forma secuencial en un lapso de 2 meses y en una proporción de 4:1 afecta más comúnmente a los hombres que a las mujeres34. El fondo de ojo suele mostrar la presencia de telangiectasias peripapilares, microangiopatía y tortuosidad vascular35. La mayor parte de los casos son causados por la mutación 11778 G>A en el genoma mitocondrial en homoplasmia36. En consecuencia, una muestra de sangre es suficiente para realizar el diagnóstico.
Neuropatía con ataxia y retinitis pigmentaria y síndrome de Leigh causado por mutaciones en el ADN mitocondrialLa neuropatía con ataxia y retinitis pigmentaria (NARP) y el síndrome de Leigh causado por mutaciones en el ADN mitocondrial, habitualmente causados por el mismo defecto en el genoma mitocondrial, representan un espectro de severidad en el grado de afectación del sistema nervioso central y periférico37. Presentan en común el habitual agravamiento o desencadenamiento de la sintomatología en asociación con una infección viral38. La presencia de retinitis pigmentaria y neuropatía sensitiva define al síndrome NARP, mientras que un compromiso más severo con afectación del sistema nervioso central (encefalomielopatía necrosante), manifestado sintomáticamente por movimientos anormales y regresión en las pautas madurativas, junto a características alteraciones en los ganglios de la base en las imágenes por RM en los primeros 3 años de la vida, definen al síndrome de Leigh39. La mayoría de los sujetos que presentan NARP y un 10-20% con síndrome de Leigh tienen mutaciones en el gen MT-ATP6 en el genoma mitocondrial, siendo el cambio en la posición 8993 el más prevalente40.
Síndrome de ataxia y neuropatía (ataxia neuropathy spectrum). Ataxia sensitiva-epilepsia mioclónicaDistintos pacientes han sido descritos en los que se combina la presencia de una neuropatía severa generalmente sensitiva con características similares a las descritas en las ganglionopatías dorsales y que sintomáticamente se expresan de manera predominante como un trastorno atáxico41 junto a una epilepsia mioclónica42 y, en muchos casos, crisis migrañosas recurrentes y disfunción hepática43. Estos síndromes de curso progresivo y herencia autosómica recesiva han recibido distintas denominaciones en el pasado. Sin embargo, considerando que comparten como etiopatogenia la presencia de mutaciones en POLG, actualmente son identificados como trastornos relacionados con POLG44,45.
Si no tiene presentación sindromática, ¿es un defecto aislado de la cadena respiratoria?Los defectos aislados de alguno de los componentes de la cadena respiratoria suelen tener una presentación clínica más agresiva y comienzo en los primeros años de vida. Habitualmente se manifiestan como una encefalomiopatía con o sin los hallazgos imagenológicos característicos del síndrome de Leigh, como una insuficiencia hepática, una miocardiopatía o la combinación de la afectación de estos sistemas46. Sin embargo, han sido descritos de forma reiterada casos que han mostrado una presentación más indolente en la edad adulta47 y que, clínicamente, manifestaban ataxias progresivas, miopatías y/o intolerancia al ejercicio48. En particular, debe ser tenido en cuenta que en aquellos pacientes con déficit primarios de CoQ10 (véase más adelante) la única manifestación sintomática puede ser la de una ataxia crónica y progresiva de comienzo más frecuentemente en la primera década de la vida, sin otros elementos clínicos que permitan diferenciarla de la extensa lista de entidades que también pueden expresarse como un trastorno crónico del equilibrio, razón por la que se recomienda la prueba supletoria terapéutica (y eventualmente diagnóstica) con coenzima Q10 en todos los pacientes con ataxias crónicas de etiologías no aclaradas49. Además, pueden resultar útiles de mención algunos elementos diferenciales de la intolerancia al ejercicio causada por disfunción mitocondrial en comparación con similar sintomatología secundaria a glucogenosis: ausencia de fenómeno de «segundo aire», mialgias sin calambres musculares, lactacidosis y reducido consumo máximo corporal de oxígeno50.
El enfoque diagnóstico de los pacientes con sospecha de déficit aislados de la cadena respiratoria debe ser dirigido a partir de la confirmación e individualización del o de los complejo/s alterado/s mediante la realización de pruebas bioquímicas funcionales en el tejido clínicamente más afectado (por ejemplo, músculo, hígado, miocardio, etc.)51. Este estudio funcional permitirá discriminar entre una de 3 posibilidades: un defecto aislado de un complejo de la cadena respiratoria; un defecto del tándem i-iii o ii-iii, y múltiples defectos de la cadena respiratoria.
Ante la presencia de un defecto aislado del complejo i deben buscarse mutaciones en los genes NDUFS1, NDUFS2, NDUFS3, NDUFS4, NDUFS5, NDUFS6, NDUFS8¸ NDUFV1 y NDUFV152. Los defectos aislados del complejo ii suelen ser consecuencia de mutaciones en los genes SDHA, SDHB, SDHC y SDHD53,54. Mutaciones en los genes TTC19, UQCRQ y BCS1L provocan disfunción aislada del complejo iii55, mientras que defectos aislados del complejo iv suelen ser consecuencia de mutaciones en SURF1, SCO1, SCO2, COX10 y COX1556. Mucho menos frecuentes son los defectos aislados del complejo v causados por mutaciones en ATP12 y TMEM7057. Cuando las pruebas funcionales indican la presencia de disfunción de los complejos i-iii o ii-iii, deben considerarse fuertemente los déficit primarios de coenzima Q10 (potencialmente reversibles con su suplementación oral), debiendo confirmarse esta sospecha diagnóstica mediante la determinación de su concentración en tejido muscular49 y la búsqueda de mutaciones en ADCK3, COQ2 y COQ858. La evidencia de múltiples defectos en la cadena respiratoria debe hacer considerar que el defecto genético más probablemente se encuentra en el genoma mitocondrial, debiendo proseguirse el algoritmo diagnóstico con la secuenciación completa del genoma mitocondrial59 o, por el contrario, de estar en presencia de múltiples deleciones del genoma mitocondrial o de depleción mitocondrial, la búsqueda molecular debe orientarse a mutaciones en los genes nucleares POLG, PEO1, TK2 y TYMP, entre otros60.
Algoritmo diagnósticoUna vez contestadas las 3 preguntas del enfoque diagnóstico presentado, puede seguirse una aproximación ordenada y secuencial de solicitud de pruebas moleculares cuyo gráfico se muestra en la figura 2 (adaptada de Wong et al.61 y McFarland et al.62) que podrá permitir la individualización del defecto molecular subyacente a la frecuentemente heterogénea y compleja presentación clínica observada. No obstante ello, en un porcentaje significativo de los casos con diagnóstico clínico-patológico de disfunción mitocondrial no podrá individualizarse el defecto molecular causante debido a que este aún es desconocido en la literatura científica o la presentación clínica es lo suficientemente atípica como para no corresponder a la sistematología propuesta61,59.
Avances en el diagnóstico molecular de los trastornos mitocondriales
Entre los avances de la genética molecular se incluye el desarrollo de las nuevas metodologías de secuenciación genómica que han posibilitado la obtención de un vasto volumen de información a un significativo menor coste63. La disponibilidad de estas nuevas tecnologías está significando un cambio paradigmático en los enfoques clásicos utilizados en la investigación y el diagnóstico de los trastornos hereditarios. Es así como resulta factible el escrutinio simultáneo de la secuencia completa de miles de genes codificantes de proteínas de la cadena respiratoria junto a la del genoma mitocondrial a partir de una única muestra biológica y a una fracción del coste que el análisis tradicional de unos pocos genes requiere64. El último tiempo mostró distintos ejemplos de la aplicación de estas metodologías en el diagnóstico de los trastornos mitocondriales. Nosotros probamos la alta sensibilidad de la pirosecuenciación masiva en la detección de mutaciones en bajos niveles de heteroplasmia en el diagnóstico de MELAS a partir de una muestra sanguínea65. Calvo et al.66 analizaron la secuencia de 1.034 genes implicados en el funcionamiento mitocondrial junto al genoma completo mitocondrial en 42 pacientes afectados de trastornos de la cadena respiratoria, pudiendo lograr la individualización diagnóstica molecular en más del 50% de los casos. Necesariamente el enfoque diagnóstico molecular de la enfermedad mitocondrial está llamado a modificarse en el futuro cercano.
FinanciaciónEste trabajo fue financiado con subsidios de investigación de CONICET, GCBA y Fundación Florencio Fiorini. Marcelo Kauffman es miembro de la carrera de investigador de CONICET y del Gobierno de la Ciudad de Buenos Aires.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
