




La b-oxidación de ácidos grasos es un proceso fisiológico crítico que permite el uso de la grasa como fuente de energía en momentos de estrés y ayuno. La deficiencia aislada de 3-hidroxiacil CoA deshidrogenasa de cadena larga (LCHADD) (OMIM#609016) es una enfermedad autosómica recesiva que afecta el metabolismo de los ácidos grasos de cadena larga y es secundaria a mutaciones en el gen HADHA. Los síntomas clínicos son heterogéneos y se desarrollan progresivamente con episodios de exacerbación, principalmente durante episodios de enfermedades intercurrentes o ayunos debido al aumento de las necesidades energéticas. Presentamos el caso de una paciente de 36 años que presentó debilidad progresiva en ambos miembros inferiores con inicio distal y progresión proximal asociada a episodios de rabdomiólisis que se desencadenan por un síndrome febril. Al examen clínico se demostró la presencia de anquilosis quirúrgica de la articulación del tobillo, debilidad en la flexión del muslo sobre la pelvis 4/5 y flexión de la pierna sobre el muslo 4/5, hipotonía muscular y atrofia muscular del compartimento posterior de la pierna, así como arreflexia de los miembros inferiores con reflejos conservados en los superiores. Ante la sospecha de una posible miopatía metabólica se solicita posología de acilcarnitinas, donde se observa aumento de C16OH, C18OH, C18:20H y C18:10H, y ante estos resultados se evalúa el gen HADHA, demostrando la presencia de la variante c.2231delT, que promueve un cambio en la matriz de lectura a partir de este momento, con la consiguiente creación de un codón de parada prematuro para la traducción de proteínas. El diagnóstico de deficiencia de LCHADD se establece demostrando una elevación de los ácidos grasos de cadena larga, especies de hidroxiacilcarnitina en el plasma y/o una mayor excreción de ácidos 3-hidroxidicarboxílicos en la orina en combinación con la identificación de variantes patógenas bialélicas en HADHA o HADHB mediante pruebas genéticas moleculares.
The b-oxidation of fatty acids is a critical physiological process that allows the use of fat as a source of energy in times of stress and fasting. Isolated long-chain 3-hydroxyacyl CoA dehydrogenase deficiency (LCHADD) (OMIM#609016) is an autosomal recessive disease that affects the metabolism of long-chain fatty acids and is secondary to mutations in the HADHA gene. Clinical symptoms are heterogeneous and develop progressively with exacerbation episodes mainly during episodes of intercurrent illness or fasting due to increased energy needs. A 36-year-old patient presented with progressive weakness in both lower limbs with distal onset and proximal progression associated with episodes of rhabdomyolysis that are triggered by a febrile syndrome. Clinical examination demonstrated the presence of surgical ankylosis of the ankle joint, weakness when flexion of the thigh over the pelvis 4/5 and flexion of the leg over the thigh 4/5, muscle hypotonia and posterior compartment muscle atrophy of the leg, areflexia of the lower limbs with preserved reflexes in the upper limbs. Given the suspicion of a possible metabolic myopathy, a dosage of acylcarnitines is requested where an increase in C16OH, C18OH, C18:20H and C18:10H is observed, and given these results, the HADHA gene is evaluated, demonstrating the presence of the variant c.2231delT, which promotes a change in the reading matrix from this point on, with the consequent creation of a premature stop codon for protein translation. The diagnosis of LCHADD deficiency is established by demonstrating an elevation of long-chain fatty acids, hydroxyacylcarnitine species in plasma, and/or increased excretion of 3-hydroxy-dicarboxylic acids in urine in combination with identification of biallelic pathogenic variants in HADHA or HADHB by molecular genetic testing.
La beta-oxidación de ácidos grasos es un proceso fisiológico crítico que permite la utilización de la grasa como fuente de energía en momentos de estrés y ayuno. La deficiencia aislada de 3-hidroxiacil CoA deshidrogenasa de cadena larga (LCHADD) (OMIM#609016) es una enfermedad autosómica recesiva que afecta el metabolismo de los ácidos grasos de cadena larga, siendo secundario a mutaciones del gen HADHA. El gen HADHA contiene 20 exones y se han descrito más de 30 variantes hasta la fecha, siendo las mutaciones homogocigotas el hallazgo más común, o con menor frecuencia la presencia de heterocigosis compuesta1.
La afectación es específica para el metabolismo de los compuestos de ácidos grasos de cadena larga C12-C16 y conduce a una acumulación de intermediarios de la beta oxidación de ácidos grasos tóxicos que causan síntomas inmediatos, así como complicaciones a largo plazo. Los síntomas clínicos son heterogéneos y se desarrollan de forma progresiva con episodios de exacerbación, principalmente durante episodios de enfermedad intercurrente o ayuno debido al aumento de las necesidades energéticas. LCHADD afecta especialmente a los órganos que necesitan de ácidos grasos de cadena larga como fuente de energía primaria, como el corazón, el músculo esquelético y el hígado2.
ObjetivosNuestro objetivo es presentar el caso de un paciente con diagnóstico de LCHADD a la edad adulta que inicia con sus síntomas en la niñez, para poner el foco en esta entidad como causa de miopatía tratable a sospechar en el paciente adulto.
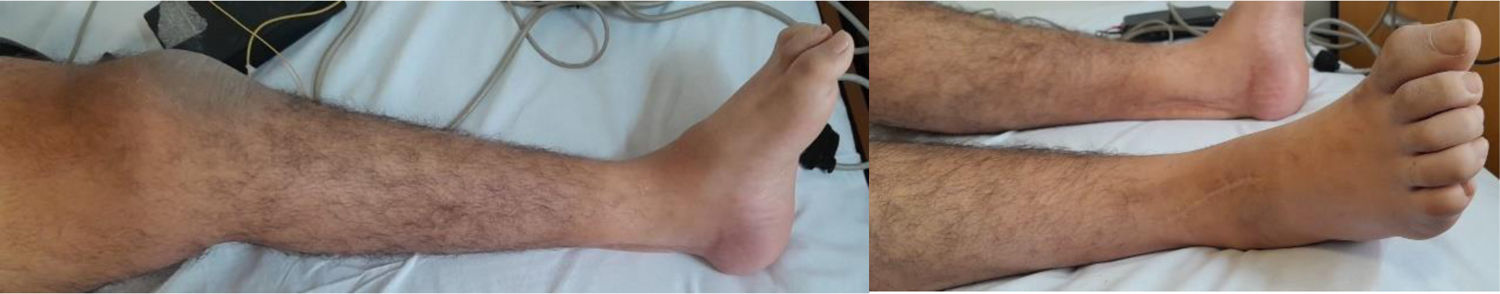
Presentación del casoPaciente de 36 años que consulta por presentar debilidad progresiva en ambos miembros inferiores de inicio distal y progresión proximal, asociada a episodios de rabdomiólisis que se desencadenan ante síndrome febril. Al examen clínico se demuestra la presencia de anquilosis quirúrgica de la articulación del tobillo, debilidad a la flexión del muslo sobre la pelvis 4/5 y de la flexión de la pierna sobre el muslo 4/5, hipotonía muscular y atrofia muscular del compartimento posterior de la pierna y arreflexia de miembros inferiores con reflejos conservados en los miembros superiores (fig. 1). Como antecedentes familiares el paciente refiere haber nacido de parto vaginal, sin antecedentes de hipoglucemia perinatales y con una marcha independiente que se consolida a los 18 meses de edad. Refiere a los 4 años de edad episodios de dolor muscular en los miembros inferiores, con claudicación de la marcha y niveles de CPK 6000, que se desencadenan ante infecciones intercurrentes, y ya a los 7 años de edad la debilidad muscular era fija, con exacerbaciones ante factores gatillantes, dificultando la marcha, por lo que le realizaron una electromiografía digital de los 4 miembros con velocidad de conducción, informándose de polineuropatía axonal, por lo que se le practicó una biopsia de músculo y nervio, donde se objetivaron cambios miopáticos (contornos poligonales, fibras anguladas atróficas estereasa positivo, atrofia neurógena con extenso agrupamiento histoquímico y fibras necróticas y en regeneración) y biopsia del nervio periférico, donde se visualizó una disminución mayor del 35% de fibras mielínicas y onion bulbs (remielinización). Por los hallazgos en la biopsia de músculo se descarta atrofia muscular espinal mediante estudio genético. A los 20 años de edad le realizan la anquilosis bilateral de la articulación del tobillo por estepaje bilateral.

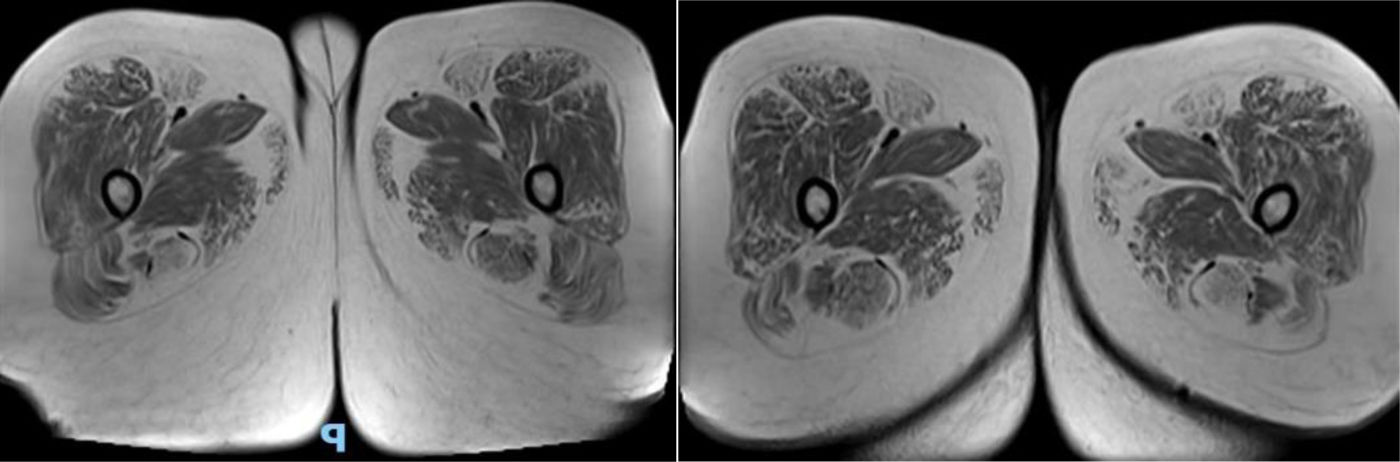
Se solicita RMI de músculos de ambos muslos, donde se visualiza en secuencia T1 la infiltración grasa de músculo sartorio, recto interno, semitendinoso y glúteo mayor bilateral (fig. 2). El estudio de laboratorio reveló una CPK de 141. La espirometría computarizada resultó con insuficiencia restrictiva de grado moderado (CVF65%3,6l y FEV169%3,62l) y la evaluación cardiológica por electrocardiograma y ecocardiograma Doppler transtorácico obtuvo resultados normales (fig. 3). La evaluación oftalmológica fue normal. Ante la sospecha de una posible miopatía metabólica se solicitó dosaje de acilcarnitinas, donde se observó un incremento de C16OH, C18OH, C18:20H y C18:10H, y ante estos resultados se procedió a la evaluación del gen HADHA que codifica la proteína trifuncional (TFP), que tiene 3 subunidades, 3-hidroxiacil-CoA deshidrogenasa de cadena larga (LCHAD), enoil-CoA hidratasa de cadena larga y tiolasa de cadena larga, que actúan en la metabolización de los ácidos grasos de cadena larga, demostrándose la presencia de la variante c.2231delT, la cual promueve un cambio de la matriz de lectura a partir de este punto, con la consecuente creación de un codón de parada prematuro de la traducción proteica (p.Phe744Serfs*7). Esta variante se demuestra en heterocigosis simple (una copia) y es descripta en la base de datos como probablemente patogénica. Según las pautas de interpretación de variantes del Colegio Americano de Genética Medica y Genómica (ACMG/AMP), los términos «variantes patógenas» y «probable variantes patógenas» son sinónimos en un entorno clínico compatible, por lo que ante la presencia de un perfil de acilcarnitina anormal, a pesar de identificarse solo una variante patogénica, se realiza el diagnóstico de deficiencia de LCHAD/TFP al no contar con la posible identificación de la enzima 3-hidroxiacilCoA deshidrogenasa de forma aislada3.
Discusión

La beta oxidación mitocondrial de ácidos grasos es esencial para la producción de energía durante el ayuno. Los síntomas de LCHADD/TFP pueden presentarse en diferentes etapas de la vida marcando según la edad de inicio y la severidad del cuadro clínico. Se describe un fenotipo severo o forma neonatal y grave, una forma infantil principalmente hepática y una forma tardía más leve que se asocia neuropatía y miopatía. Los síntomas suelen ir empeorando por el ayuno prolongado y/o glucosa insuficiente en relación con el aumento de la demanda durante una enfermedad aguda. Entre los signos clínicos la hipoglucemia hipocetósica es un clásico de todo trastorno de la beta oxidación de ácidos grasos, asociada a miocardiopatía e insuficiencia hepática2,3.
Existe un fenotipo severo o neonatal que se manifiesta generalmente a los pocos días de nacer, con un síndrome similar al de Reye, con encefalopatía, hipoglucemia hipocetósica, hepatomegalia y acidosis láctica. La mortalidad en estos casos es elevada y es secundaria a la descompensación cardiaca por el desarrollo de una miocardiopatía dilatada con arritmias e insuficiencia cardiaca y también a la insuficiencia hepática. Puede presentarse un fenotipo intermedio o infantil después de los 2 años de edad con síntomas de hipoglucemia hipocetósica precipitada por una infección o ayuno asociado a acidosis láctica. Durante las crisis, al examen se constata debilidad muscular, dificultad en la alimentación e hipotonía. Se demuestran enzimas hepáticas y creatinfosfocinasa (CPK) elevadas, y con la evolución del cuadro y las crisis reiteradas se puede desarrollar miocardiopatía dilatada o hipertrófica, cirrosis hepática y retrasos del crecimiento. Los pacientes con fenotipos leves o adolescentes desarrollan síntomas neuromusculares presentando una miopatía asociada a rabdomiólisis desencadenada por el ejercicio, exposición al frío, ayuno o infección. Estos episodios se asocian a dolor muscular difuso, debilidad muscular aguda, mioglobinuria y elevación de la CPK más de 5 veces el límite superior de la normalidad. Las complicaciones a largo plazo incluyen polineuropatía de características axonal y sensitivo motora, sin embargo puede ser puramente sensitiva o motora. Dentro de las complicaciones a largo plazo la retinopatía y la neuropatía periférica son típicas de LCHADD/TFP y no se presentan en otros trastornos de la beta oxidación de ácidos grasos mitocondriales4.
La resonancia magnética muscular muestra patrones de cambios prominentes en STIR que reflejan rabdomiólisis y aumento de la intensidad de la señal en T1 que refleja la infiltración grasa del tejido muscular3.
El diagnóstico de deficiencia de LCHADD se establece al demostrar una elevación de hidroxiacilcarnitina en plasma y/o aumento de excreción de ácidos 3-hidroxi-dicarboxílicos en orina en combinación con identificación de variantes patogénicas bialélicas en HADHA o HADHB por pruebas de genética molecular. Según las pautas de interpretación de variantes de ACMG/AMP, términos «variantes patogénicas» y «probables variantes patogénicas» son sinónimos en un entorno clínico, lo que significa que ambas se consideran diagnósticas. Se concluye que en un individuo con un perfil de acilcarnitina anormal y cuadro clínico compatible tienen como diagnóstico LCHADD incluso si solo se identifica una sola variante patogénica o probablemente patogénica en el estudio molecular, como ocurrió en el paciente expuesto3–5. Es para destacar que no se realizó el análisis de alteraciones en regiones no codificantes del genoma, tales como regiones reguladoras, secuencias intergénicas e intrónicas distantes de los exomas por técnicas como Sanger o reacción de cadena de la polimerasa, ni tampoco el estudio genético de los progenitores, por lo que no podemos descartar que el paciente presente una heterocigosis compuesta.
Las correlaciones genotipo-fenotipo son difíciles de establecer en presencia de heterocigosis compuesta, no así en caso de homocigosis, como por ejemplo en la variante c-1528G>C (mutación más frecuente) o c.985A>G donde los pacientes experimentan mayor compromiso miocárdico y alteración de la conciencia, pero menos ataques de rabdomiólisis6. Diferentes autores sugieren que el nivel de actividad enzimática residual estaría en relación con el inicio de los síntomas, un nivel de actividad<10% presenta un riesgo de aparición sintomática de la enfermedad más temprana y pacientes con>20% de actividad pueden permanecer asintomáticos a lo largo de la vida. Los factores ambientales que actúan como gatillantes (infecciones, exposición a fármacos y ayuno) confunden aún más las correlaciones entre genotipo y fenotipo7.
El manejo dietético es el estándar actual de atención, con directrices que recomiendan evitar el ayuno, una dieta baja en grasas y alta en hidratos de carbono y el consumo de triglicéridos de cadena media (TCM) o triheptanoína (Dojolvi®) para evitar el defecto en el metabolismo de los ácidos grasos de cadena larga. Dietistas expertos evalúan la energía de cada paciente y las necesidades de nutrientes en función de la edad, el sexo, el peso y el nivel de actividad y las manifestaciones clínicas. La ingesta de calorías proporcionada por proteínas, hidratos de carbono y grasas son monitoreados y ajustados según sea necesario para satisfacer las necesidades cambiantes del paciente. A pesar de la adherencia total a la dieta y la atención óptima, la mayoría de los pacientes con LCHADD experimentan hospitalizaciones frecuentes y afectación de órganos de forma progresiva. En el caso de descompensaciones leves el tratamiento puede ser ambulatorio, indicando la disminución del intervalo de ayuno, la administración de antipiréticos para la fiebre y antieméticos según sea necesario para los vómitos. El tratamiento de las descompensaciones severas incluye la hospitalización con la administración de suero intravenoso que contenga al menos un 10% de dextrosa y terapia con bicarbonato para la acidosis metabólica grave, manejo de hiperamonemia, rabdomiólisis y de la posible miocardiopatía asociada5,6.
Como suplemento dietético el aceite TCM no está regulado en la misma medida que los medicamentos por los controles de fabricación, por lo tanto, cada aceite TCM comercial puede variar en la composición de los componentes de triglicéridos específicos. Aunque se plantea la hipótesis de que el componente más utilizado por los pacientes con LCHADD es el C7, el porcentaje de C7 identificado en las formulaciones de aceite TCM comúnmente disponibles puede oscilar entre el 40% y el 70%. Además, los pacientes con LCHADD parecen tener diferentes niveles de adherencia y/o tolerabilidad al aceite TCM, independientemente de la gravedad de la enfermedad, lo que agrava cualquier riesgo subyacente de catabolismo y crisis metabólicas. En función del reconocimiento de este déficit, se propuso la triheptanoína, un heptanoil-triglicérido suplementario de calidad alimentaria, como un TCM fisiológicamente más equilibrado. Para prevenir complicaciones tanto agudas como crónicas, anaplerosis (producción de sustratos energéticos) y cataplerosis (catabolismo de sustratos energéticos) deben estar en equilibrio para la homeostasis de energía. La triheptanoína puede equilibrar la cataplerosis aumentando la producción de energía mediante el suministro de sustratos anapleróticos y ATP para apoyar la gluconeogénesis y la glucogénesis. Se ha demostrado que es eficaz en las complicaciones a corto y largo plazo y muchas publicaciones han informado de que la triheptanoína proporciona una disminución significativa en los principales eventos clínicos, tales como episodios de rabdomiólisis. El régimen con trihepatonoína debe adecuarse de acuerdo con la edad, el peso ideal y la actividad y se recomienda que constituya el 25-35% de la ingesta calórica diaria. Se recomienda suspender las formulaciones con TCM en pacientes que harán la transición a la triheptanoína. El sobrepeso es más común en pacientes con LCHADD en comparación con la población general. Las tasas de sobrepeso y obesidad se reportaron como 60% y 10-30%, respectivamente. La alimentación frecuente, evitar el hambre, una dieta baja en grasas y alta en hidratos de carbono y una baja capacidad de ejercicio pueden resultar en sobrepeso y obesidad iatrogénicos, pero la triheptanoína no está asociada con aumento de peso4-10.
ConclusionesLa deficiencia de hidroxiacil-CoA deshidrogenasa de cadena larga (LCHADD) típicamente se presenta con un fenotipo severo o grave, demostrándose a los pocos días de nacer con hipoglucemia, hepatomegalia, encefalopatía y, a menudo, cardiomiopatía, o por un fenotipo intermedio que se caracteriza por hipoglucemia hipocetósica precipitada por infección o ayuno en la infancia. Las presentaciones clínicas de LCHADD, donde los síntomas principales se caracterizan por miopatía y/o neuropatía, son menos frecuentes, por lo que creemos que es un diagnóstico a tener en cuenta en pacientes con miopatía e historia de rabdomiólisis, siendo mucho más sugestivo el diagnóstico si se demuestra la presencia de una polineuropatía, ya que existen tratamientos disponibles que cambian la evolución natural de la misma. Este paciente presenta como dato de interés la presencia de una sola mutación en el estudio genético, lo que no invalida el diagnóstico, ya que se asocia a un cuadro clínico y perfil de acilcarnitinas compatible con el diagnóstico11.
Conflicto de interesesLos autores declaran no tener conflicto de intereses.
