



Las ataxias cerebelosas autosómicas recesivas (ACAR) constituyen un grupo muy heterogéneo de trastornos neurodegenerativos que puede presentarse como síndrome cerebeloso puro o complejo, asociado a otras manifestaciones neurológicas o sistémicas que pueden servir de ayuda en su complejo abordaje diagnóstico1,2. En los últimos años, el estudio genético ha cobrado mayor importancia para esclarecer la etiología de determinadas ACAR3.
Presentamos el caso de una fratría de 2 hermanos varones de 27 y 20 años con retraso psicomotor y trastorno del lenguaje. Ambos son hijos de padres consanguíneos. No existen antecedentes de enfermedades neurológicas en la familia. El embarazo, parto, puerperio, así como el período neonatal y la infancia precoz transcurrieron sin incidencias. Los cribados endocrinometabólicos y auditivos iniciales habituales fueron normales. El mayor de los hermanos, «A», presentó un desarrollo psicomotor normal hasta los 7 años, cuando se evidenció un retraso del aprendizaje y de la motricidad (CI: 57), con alteración del lenguaje a partir de los 17 años. El menor de los hermanos, «P», presentó un retraso psicomotor evidente a partir de los 5 años, con afectación tanto del lenguaje como de la motricidad fina (CI: 47). Ambos hermanos mantienen la deambulación autónoma actualmente. En la exploración neurológica de «A» se evidenciaba un estrabismo convergente en el ojo derecho (por paresia del VI par craneal derecho) junto con un síndrome cerebeloso bilateral asimétrico (disartria escandida y dismetría apendicular, con marcha atáxica y tándem imposible), así como signos de afectación piramidal incipiente (reflejos osteotendinosos vivos con reflejo cutáneo plantar indiferente bilateral). En el caso de «P» el síndrome cerebeloso no era tan florido (disartria escandida y nistagmo en levoversión), siendo más llamativa la torpeza manipulativa y la imposibilidad para realizar la marcha en tándem, sin signos de afectación de la vía piramidal. Se realizó una valoración oftalmológica de ambos pacientes, que no mostró hallazgos patológicos. El perímetro cefálico ajustado a sexo, edad, peso y estatura de los 2 hermanos fue normal (56,2cm en «A» y 56,8cm en «P»).
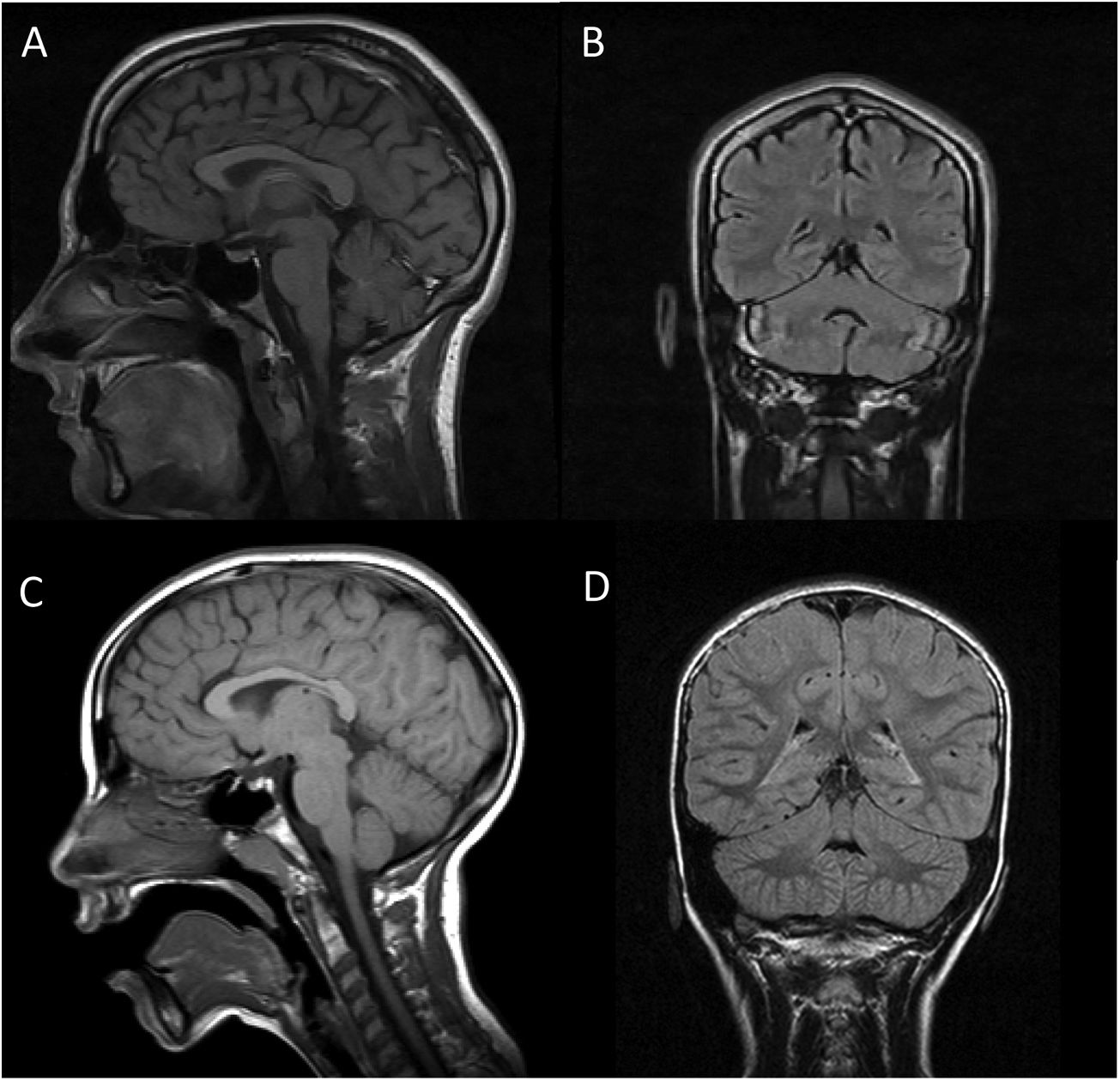
Se completó en ambos casos el estudio diagnóstico con resonancia magnética cerebral (fig. 1) y muscular, electroencefalograma, electromiograma-electroneurograma y potenciales evocados auditivos, con resultado normal (salvo quizá una mínima atrofia vermiana en «A»). Se realizó el despistaje analítico de aquellas ACAR potencialmente susceptibles de iniciar tratamiento modificador, siendo el resultado también normal. Además, se inició un estudio genético básico con cariotipo, determinación de síndrome de X frágil y array-CGH 60K, todo ello sin alteraciones. Finalmente, se realizó un panel de genes de ataxia, determinándose que tanto «A» como «P» son portadores en homocigosis de la variante NM_016955.3: c.1321G>A; p.(Gly441Arg) en el exón 11 del gen SEPSECS, siendo sus padres portadores en heterocigosis; dicha variante ha sido considerada como patógena por varios programas de predicción in silico, como refieren van Dijk et al.4. Se trata de una mutación missense descrita con anterioridad en un caso de una paciente con microcefalia e hipoplasia cerebelosa progresiva4.
 y FLAIR coronal (imágenes B y D). Las imágenes A y B corresponden al paciente A, mientras que C y D corresponden al paciente P. En ambos casos se objetiva la normalidad de las estructuras troncoencefálicas y cerebelosas, sin signos típicos de hipoplasia pontocerebelosa salvo quizá una mínima atrofia vermiana en el paciente A (imágenes A y B).")
RM cerebral en secuencias T1 corte sagital (imágenes A y C) y FLAIR coronal (imágenes B y D). Las imágenes A y B corresponden al paciente A, mientras que C y D corresponden al paciente P. En ambos casos se objetiva la normalidad de las estructuras troncoencefálicas y cerebelosas, sin signos típicos de hipoplasia pontocerebelosa salvo quizá una mínima atrofia vermiana en el paciente A (imágenes A y B).
Variantes patogénicas en el gen SEPSECS se han asociado con hipoplasia pontocerebelosa tipo 2D, un trastorno neurodegenerativo de inicio prenatal o infantil caracterizado por una atrofia grave de puente y cerebelo que cursa con ataxia, corea, hipotonía, nistagmo, microcefalia, espasticidad y discapacidad cognitiva, con atrofia tanto cerebral como cerebelosa progresivas4–6. Recientemente se ha ampliado el fenotipo clínico, describiéndose formas más leves y de inicio más tardío (milder); se hipotetiza que mutaciones missense, como la de nuestros pacientes, podrían resultar en una mayor actividad residual de la proteína SepSecS, lo cual explicaría el inicio más tardío y la menor gravedad clínica4.
El gen SEPSECS se encuentra en el brazo corto del cromosoma 4 (4p15.2), y su función es catalizar el último paso en la vía de la síntesis de la selenocisteína (Sec). En concreto, este gen codifica la proteína SepSecS, que da lugar a la enzima O-fosfoseril-ARNt:selenocisteinil-ARNt sintetasa, la cual es clave en la única ruta biosintética de Sec en eucariotas y arqueas7. La Sec es una selenoproteína especialmente relevante en el desarrollo del cerebro de los mamíferos7.
El fenotipo clínico de las ACAR, a menudo se solapa y presenta una gran variabilidad. Así, lo prioritario en el abordaje diagnóstico inicial debe ser descartar aquellas enfermedades que tienen tratamiento eficaz, de cara a iniciarlo lo más tempranamente posible2. Dentro de este grupo se incluyen las ataxias recesivas debidas a defectos metabólicos congénitos (abetalipoproteinemia, ataxia por déficit de vitamina E, etc.)2. Descartadas aquéllas, se debe ampliar el diagnóstico en base al fenotipo, teniendo en cuenta en primer lugar los cuadros más frecuentes (ataxia de Friedreich, entre otras). Tal y como se recoge en las últimas recomendaciones1, el siguiente paso será realizar el estudio genético, primero con el estudio de genes aislados (p. ej., expansión del gen FXN en ataxia de Friedreich), seguido de técnicas más complejas de next generation sequencing (NGS), incluyendo paneles genéticos, whole exome sequencing (WES) e incluso whole genome sequencing (WGS).
En resumen, presentamos un caso de una fratría de 2 hermanos con ataxia cerebelosa progresiva sin atrofia en la neuroimagen debida a una mutación en el gen SEPSECS, hasta ahora únicamente descrita como causante de hipoplasia pontocerebelosa tipo 2D.
FinanciaciónEl presente manuscrito no ha recibido financiación alguna para su elaboración.

