



It has been observed in recent years that levels of such molecules as calcitonin gene–related peptide (CGRP) and, to a lesser extent, the pituitary adenylate cyclase–activating peptide are elevated during migraine attacks and in chronic migraine, both in the cerebrospinal fluid and in the serum. Pharmacological reduction of these proteins is clinically significant, with an improvement in patients’ migraines. It therefore seems logical that one of the main lines of migraine research should be based on the role of CGRP in the pathophysiology of this entity.
DevelopmentThe Spanish Society of Neurology’s Headache Study Group decided to draft this document in order to address the evidence on such important issues as the role of CGRP in the pathophysiology of migraine and the mechanism of action of monoclonal antibodies and gepants; and to critically analyse the results of different studies and the profile of patients eligible for treatment with monoclonal antibodies, and the impact in terms of pharmacoeconomics.
ConclusionsThe clinical development of gepants, which are CGRP antagonists, for the acute treatment of migraine attacks, and CGRP ligand and receptor monoclonal antibodies offer promising results for these patients.
En los últimos años se ha observado que moléculas como el péptido relacionado con el gen de la calcitonina (CGRP) y, en menor grado, el péptido activador de la adenilato-ciclasa pituitaria (PACAP) estaban elevadas durante los ataques de migraña y en la migraña crónica tanto en líquido cefalorraquídeo como en suero y que su reducción farmacológica tenía una significación clínica con una mejoría en la migraña de los pacientes. Es lógico por tanto que una de las principales líneas de investigación en migraña se base en el papel del CGRP en la fisiopatología de esta entidad.
DesarrolloDesde el Grupo de Estudio de Cefaleas de la Sociedad Española de Neurología nos planteamos la redacción de este documento, cuyo objetivo es abordar, basándonos en la evidencia publicada, cuestiones tan importantes como el papel del CGRP en la fisiopatología de la migraña, el mecanismo de acción de los AMC y de los gepantes, el análisis crítico de los resultados de los diferentes estudios, el perfil del paciente que podría ser candidato al tratamiento con AMC y su impacto en términos de farmacoeconomía.
ConclusionesEl desarrollo clínico de los gepantes, antagonistas del CGRP, para el tratamiento agudo del ataque de migraña y de los anticuerpos monoclonales (AMC) contra ligando y contra el receptor del CGRP, ofrecen resultados esperanzadores para nuestros pacientes.
In recent years, one of the main lines of research in migraine has been the search for biomarkers that may contribute to our understanding of the pathophysiology of the condition and to the design of new treatment options, leading to the advent of what is known as “precision medicine in migraine.” Elevated levels of such molecules as calcitonin gene–related peptide (CGRP) and, to a lesser extent, pituitary adenylate cyclase–activating polypeptide (PACAP) have been detected during migraine attacks1 and in chronic migraine (CM), both in the cerebrospinal fluid2 and in the serum3; pharmacological treatments to reduce their levels are associated with a clinically significant improvement in migraine.4,5 The clinical development of gepants, CGRP antagonists, for the treatment of migraine attacks was temporarily interrupted due to transaminitis,6 although current results are promising.7 Subsequently, monoclonal antibodies were developed that target the CGRP ligand and receptor and do not interact with liver enzymes; these molecules are large (preventing them from crossing the blood-brain barrier [BBB]) and present a high level of specificity. Recently completed clinical trials with anti-CGRP monoclonal antibodies in episodic migraine (EM) and CM, and with gepants to treat attacks, offer promising results for our patients.
This study by the Spanish Society of Neurology’s Headache Study Group (GECSEN) reviews the published evidence to address such important issues as the role of CGRP in the pathophysiology of migraine, the action mechanisms of monoclonal antibodies and gepants, the critical analysis of the results of different studies, the profile of potential candidates for treatment with monoclonal antibodies, and the impact of these drugs in terms of pharmacoeconomics.
MethodsWe established an expert committee comprising 16 GECSEN members, who independently submitted their recommendations, presented in a question and answer format, on a series of highly important issues related to the new treatments targeting CGRP. These recommendations are based on the available evidence and on clinical experience. Consensus was reached according to the Delphi method.
Following this methodology, the most clinically relevant subjects are addressed by the following questions:
- 1
What is the role of CGRP in the pathophysiology of migraine?
- 2
Is CGRP a biomarker of migraine?
- 3
What is the role of gepants in the symptomatic treatment of migraine?
- 4
What monoclonal antibodies have been tested in clinical trials of migraine? What is the efficacy and safety of these drugs?
- 5
What are the benefits of monoclonal antibodies over current treatments for episodic and chronic migraine?
- 6
What are the characteristics of patients who respond to anti-CGRP monoclonal antibodies?
- 7
When is migraine considered to be treatment-resistant?
- 8
In terms of pharmacoeconomics, what do anti-CGRP monoclonal antibodies contribute to the treatment of migraine?
In the last decade, advances in basic research and various functional neuroimaging studies have improved our understanding of the pathophysiology of such a complicated disease as migraine.
The trigeminovascular system plays a central role in pain during migraine attacks. It is composed of the vessels of the dura mater and pia mater and sensory afferents from the ophthalmic branch of the trigeminal nerve, which surround these vessels (Aδ and C polymodal nociceptive afferents). These fibres transmit nociceptive information to the trigeminal nucleus caudalis, which is part of the trigeminocervical complex and receives afferents from the first cervical roots.
Two potentially compatible theories have been proposed to explain the activation of the trigeminovascular system during migraine attacks. Cortical spreading depression, the pathophysiological substrate of migraine aura, is a wave of neuronal and glial depolarisation that moves across the cerebral cortex at a rate of 2.5-5 mm/second and is followed by a sustained suppression of spontaneous neuronal activity. It is accompanied by changes in vascular calibre and blood flow (an initial phase of cortical hyperaemia, lasting several minutes, followed by a more prolonged phase of hypoperfusion) and changes in energy metabolism, leading to the release of chemical mediators (prostaglandins and such excitatory neurotransmitters as glutamate and nitric oxide) into the extra- and perivascular spaces. Cortical changes also occur during this process; these include the release of adenosine triphosphate and glutamate from neurons and glia and the activation of metalloproteases that rupture the BBB, allowing chemical mediators to activate trigeminal terminals surrounding the meningeal vessels.8
Pain seems to be generated in the brainstem nuclei, locus coeruleus, and dorsal raphe nucleus. This theory is based on neuroimaging studies, which have demonstrated that these structures are activated in this phase of migraine; however, limbic system structures are also activated (prefrontal cortex, cingulate, insula, thalamus). Furthermore, these nuclei, and especially the periaqueductal grey matter, participate in the descending antinociceptive system, and can therefore modulate pain signals. Their activation results in dysfunction and promotes nociceptive afferent inputs.9
In both cases, activation of the trigeminovascular system is the final result. This activation occurs bidirectionally, and has 2 consequences: orthodromic conduction, with nociceptive information being transmitted towards the trigeminal nucleus caudalis, thalamus, and somatosensory cortex; and antidromic conduction, causing aseptic meningeal inflammation secondary to the release of vasoactive neuropeptides (substance P, CGRP, VIP, endothelin-3, PACAP, neuropeptide Y, peptide histidine-methionine, and neurokinin A).
CGRP is the molecule that has most consistently been associated with activation of the trigeminovascular system. It is one of 6 peptides in the calcitonin family; the α subform is highly abundant in the cerebral blood vessels and in the trigeminal ganglion. It has a potent vasodilatory effect and facilitates nociception. CGRP and its receptor are also known to be expressed in such other locations as afferent terminations from the trigeminal nucleus caudalis (hence also playing a role in central sensitisation), cerebellum, dura mater, periaqueductal grey matter, thalamus, hypothalamus, limbic system, and cortex. There is also extensive evidence of its involvement in the phenomenon of central sensitisation and, therefore, in the allodynia and hyperalgesia reported by some patients during migraine attacks.10–14 Finally, CGRP is reported to play a role in photophobia, one of the most characteristic features of migraine attacks.
Is CGRP a biomarker of migraine?Perhaps we should first consider whether there is a need for a biomarker for an entity like migraine, whose clinical diagnosis appears to be straightforward. The answer to this question is a definite yes, based on 2 arguments. Firstly, misdiagnosis occurs, especially in patients with CM.15 Secondly, having a reliable biomarker enables objective evaluation of treatment response. Furthermore, in such a condition as migraine, which is invisible, the disease gains recognition as a biological entity, reducing stigma.
In migraine attacks, pain is caused by activation of the trigeminovascular system, composed of afferents from the trigeminal nerve and efferents from the parasympathetic fibres of the facial nerve. It appears reasonable that biomarkers of migraine may include some of the more than 10 neuropeptides involved in activation of this system, affecting either the trigeminal nerve (CGRP, substance P, neurokinin A, amylin, cholecystokinin-8, S100B) or the parasympathetic nervous system (VIP, PACAP, heliodermin, helospectin I and II, peptide histidine-methionine, nitric oxide).
In 1994, Lars Edvinsson and Peter Goadsby1 were able to measure the levels of several of these peptides in blood samples taken from the external jugular vein ipsilateral to pain during a migraine attack. The only peptide consistently observed at elevated levels was CGRP. Furthermore, CGRP normalised after treatment with sumatriptan16; a more recent study found this also to occur after treatment with rizatriptan.17 However, levels of substance P, thought at the time to play a crucial role, were not elevated, and VIP levels were only increased in patients with parasympathetic autonomic symptoms. The need to collect blood from the jugular vein limits the reproducibility of these findings. However, in a 2012 study with ELISA techniques, Rodríguez-Osorio et al.18 detected elevated levels of CGRP in peripheral blood during migraine attacks.18 The importance of CGRP was once more demonstrated when experimental studies showed that intravenous injection of the neuropeptide was able to induce delayed migraine-like attacks only in patients diagnosed with migraine.19 These results confirmed the role of CGRP as a potential biomarker of the acute phase of migraine.
Subsequently, studies using animal models found that in addition to its involvement in the acute phase of migraine, CGRP also participates in the phenomenon of sensitisation and in migraine transformation.20 Therefore, it is unsurprising that elevated CGRP levels should be detected during attacks in the peripheral blood of patients with CM.3 Other studies have shown that the saliva and cerebrospinal fluid also present increased CGRP levels during migraine attacks in patients with EM and in the interictal phase in CM.2 However, these findings have been questioned due to methodological problems with sample processing.
A biomarker must be capable of predicting the response to specific treatment and must normalise with treatment (as is the case with glycaemia in diabetes, for example). CGRP meets both requirements reasonably well in CM: high levels of the peptide can predict good response to treatment with onabotulinumtoxinA (OnabotA), and CGRP levels have been shown to decrease following proper pericranial injection of the drug.4,5,21
The available data may seem to suggest that peripheral blood CGRP is the first biomarker of migraine, both for acute pain and for migraine transformation. Unfortunately, we are far from being able to categorically support this hypothesis. Approximately one-third of patients with migraine present similar CGRP levels to those of controls without headache, both during attacks and in the interictal phase of CM. This is a crucial point, as it indicates that CGRP may not be the most relevant factor in some cases. In fact, as reported by Edvinsson and Goadsby,1 levels of such parasympathetic peptides as VIP also increase during migraine attacks and in CM.22 This would explain why the results of acute treatment with gepants or anti-CGRP monoclonal antibodies have not been as spectacular as we may expect if CGRP was the only molecule involved. Furthermore, it has not been confirmed that CGRP in the jugular vein is produced as a result of trigeminal activation, despite data from some experimental studies suggesting that this is indeed the case.23 Another important issue is the reproducibility and reliability of the ELISA tests available on the market. Finally, the influence of these peptides’ short half-life on the results of studies from different laboratories is unknown.
In conclusion, there is an undeniable need for a peripheral blood biomarker of migraine, and CGRP seems to be the most suitable. However, we are far from being able to affirm its status as a reliable, reproducible biomarker of migraine in clinical practice.
What is the role of gepants in the symptomatic treatment of migraine?Gepants, CGRP antagonists, represent a new class of treatment for the symptomatic management of migraine, and an alternative to triptans, which target the 5-HT1B and 5-HT1D receptors.24,25 Double-blind randomised controlled trials have shown that gepants are superior to placebo in reducing pain intensity, presenting similar efficacy to triptans but a better safety profile.
Olcegepant (BIBN 4096BS) was the first gepant found to be more efficacious than placebo in reducing pain intensity, showing promising results in the rate of recurrence at 2 hours, persistence of response over the next 24 hours, and improvements in associated symptoms including nausea, photophobia, and phonophobia. The most frequent adverse reaction was paraesthesia.26,27 The drug was administered intravenously, making it an impractical choice.
The development of telcagepant (MK-0974) was suspended, despite good results in comparison to rizatriptan 10 mg in a double-blind randomised controlled trial.28 The reason for this was that a subsequent study assessing the preventive treatment of migraine with daily administration of the drug over 3 months, compared against placebo, reported several cases of hepatotoxicity.6 Rimegepant (BMS-927711) has been shown to be more efficacious than placebo for parameters including absence of pain at 2 hours in a phase 2 trial, with similar figures to those reported for sumatriptan 100 mg. A phase 3 trial (NCT03235479) is currently underway.29
Finally, phase 2a trials of ubrogepant (MK-1602) have shown the drug to be more efficacious than placebo, with a low rate of adverse reactions. Two phase 3 trials (NCT02828020 and NCT02867709) have recently been completed, and the open-label extension phase (NCT02873221) to assess its safety and tolerability remains active.7
Gepants may present similar efficacy to that of the more potent triptans. Their advantages reside in the longer duration of their effect (resulting in a pharmacoeconomical advantage, with fewer doses being needed), their better tolerability, and especially the lack of cardiovascular secondary effects. The main disadvantage of these drugs when compared against some triptans is their ability to cross the BBB, causing central nervous system adverse reactions (somnolence).
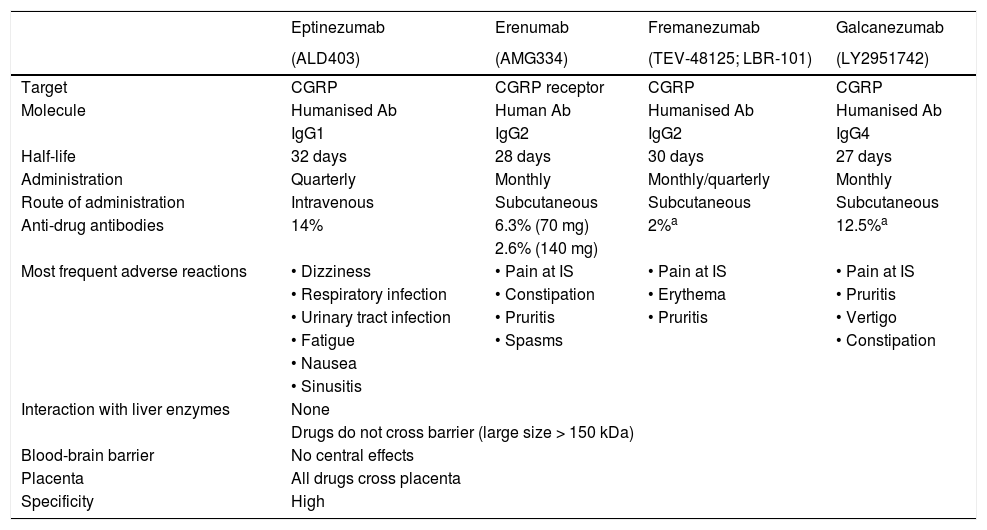
What monoclonal antibodies have been tested in clinical trials of migraine? What is the efficacy and safety of these drugs?Four monoclonal antibodies are currently in the clinical development phase: erenumab, fremanezumab, galcanezumab, and eptinezumab. Table 1 lists the most relevant characteristics. Eight phase 3 clinical trials including patients with EM and CM have been published to date (Table 2), although more trials are underway, with results to be published soon.
Characteristics of monoclonal antibodies for migraine.
| Eptinezumab | Erenumab | Fremanezumab | Galcanezumab | |
|---|---|---|---|---|
| (ALD403) | (AMG334) | (TEV-48125; LBR-101) | (LY2951742) | |
| Target | CGRP | CGRP receptor | CGRP | CGRP |
| Molecule | Humanised Ab | Human Ab | Humanised Ab | Humanised Ab |
| IgG1 | IgG2 | IgG2 | IgG4 | |
| Half-life | 32 days | 28 days | 30 days | 27 days |
| Administration | Quarterly | Monthly | Monthly/quarterly | Monthly |
| Route of administration | Intravenous | Subcutaneous | Subcutaneous | Subcutaneous |
| Anti-drug antibodies | 14% | 6.3% (70 mg) | 2%a | 12.5%a |
| 2.6% (140 mg) | ||||
| Most frequent adverse reactions | • Dizziness | • Pain at IS | • Pain at IS | • Pain at IS |
| • Respiratory infection | • Constipation | • Erythema | • Pruritis | |
| • Urinary tract infection | • Pruritis | • Pruritis | • Vertigo | |
| • Fatigue | • Spasms | • Constipation | ||
| • Nausea | ||||
| • Sinusitis | ||||
| Interaction with liver enzymes | None | |||
| Drugs do not cross barrier (large size > 150 kDa) | ||||
| Blood-brain barrier | No central effects | |||
| Placenta | All drugs cross placenta | |||
| Specificity | High | |||
Ab: antibody; CGRP: calcitonin gene–related peptide; Ig: immunoglobulin; IS: injection site; kDa: kilodalton.
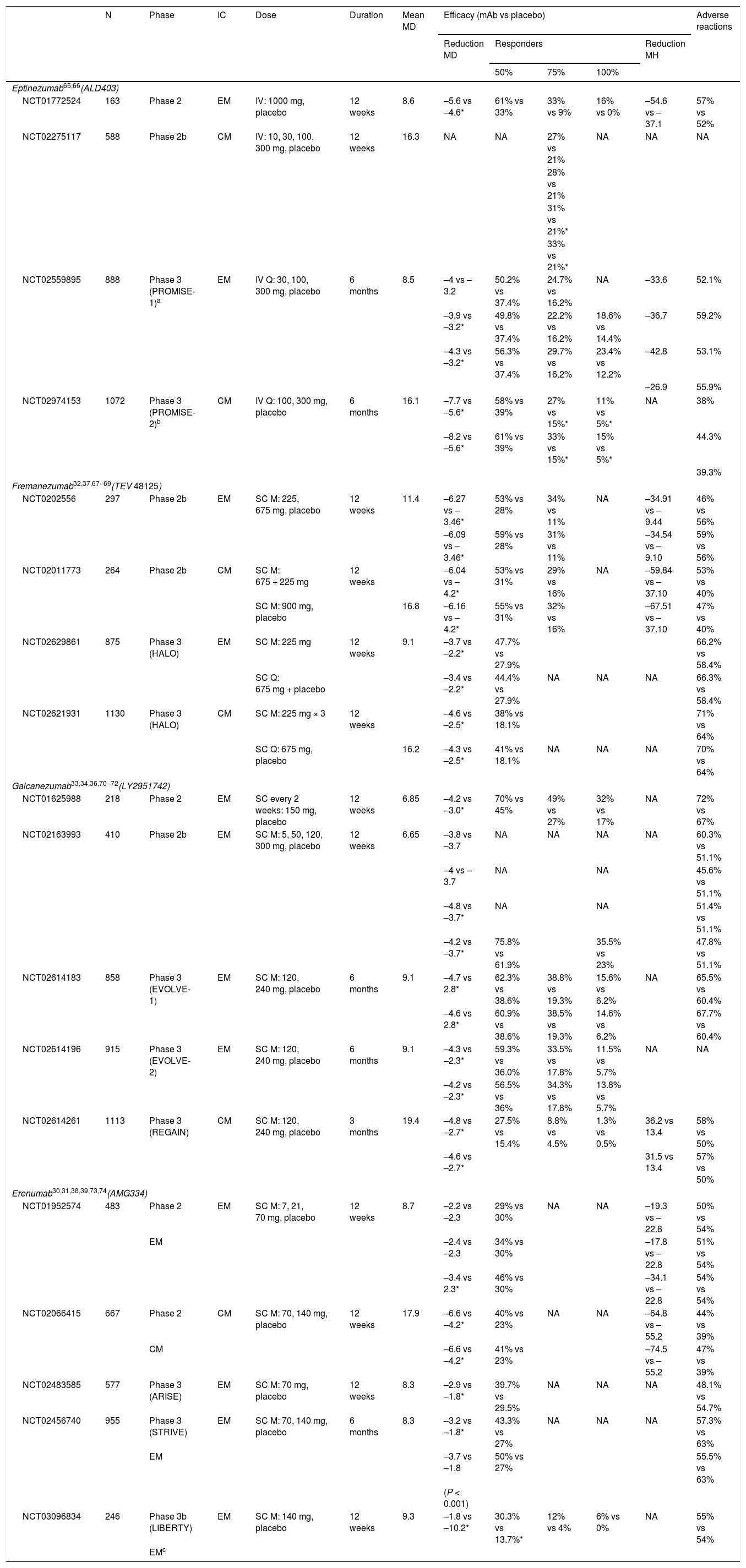
Clinical trials of monoclonal antibodies for migraine.
| N | Phase | IC | Dose | Duration | Mean MD | Efficacy (mAb vs placebo) | Adverse reactions | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reduction MD | Responders | Reduction MH | ||||||||||
| 50% | 75% | 100% | ||||||||||
| Eptinezumab65,66(ALD403) | ||||||||||||
| NCT01772524 | 163 | Phase 2 | EM | IV: 1000 mg, placebo | 12 weeks | 8.6 | –5.6 vs –4.6* | 61% vs 33% | 33% vs 9% | 16% vs 0% | –54.6 vs –37.1 | 57% vs 52% |
| NCT02275117 | 588 | Phase 2b | CM | IV: 10, 30, 100, 300 mg, placebo | 12 weeks | 16.3 | NA | NA | 27% vs 21% | NA | NA | NA |
| 28% vs 21% | ||||||||||||
| 31% vs 21%* | ||||||||||||
| 33% vs 21%* | ||||||||||||
| NCT02559895 | 888 | Phase 3 (PROMISE-1)a | EM | IV Q: 30, 100, 300 mg, placebo | 6 months | 8.5 | –4 vs –3.2 | 50.2% vs 37.4% | 24.7% vs 16.2% | NA | –33.6 | 52.1% |
| –3.9 vs –3.2* | 49.8% vs 37.4% | 22.2% vs 16.2% | 18.6% vs 14.4% | –36.7 | 59.2% | |||||||
| –4.3 vs –3.2* | 56.3% vs 37.4% | 29.7% vs 16.2% | 23.4% vs 12.2% | –42.8 | 53.1% | |||||||
| –26.9 | 55.9% | |||||||||||
| NCT02974153 | 1072 | Phase 3 (PROMISE-2)b | CM | IV Q: 100, 300 mg, placebo | 6 months | 16.1 | –7.7 vs –5.6* | 58% vs 39% | 27% vs 15%* | 11% vs 5%* | NA | 38% |
| –8.2 vs –5.6* | 61% vs 39% | 33% vs 15%* | 15% vs 5%* | 44.3% | ||||||||
| 39.3% | ||||||||||||
| Fremanezumab32,37,67–69(TEV 48125) | ||||||||||||
| NCT0202556 | 297 | Phase 2b | EM | SC M: 225, 675 mg, placebo | 12 weeks | 11.4 | –6.27 vs –3.46* | 53% vs 28% | 34% vs 11% | NA | –34.91 vs –9.44 | 46% vs 56% |
| –6.09 vs –3.46* | 59% vs 28% | 31% vs 11% | –34.54 vs –9.10 | 59% vs 56% | ||||||||
| NCT02011773 | 264 | Phase 2b | CM | SC M: 675 + 225 mg | 12 weeks | –6.04 vs –4.2* | 53% vs 31% | 29% vs 16% | NA | –59.84 vs –37.10 | 53% vs 40% | |
| SC M: 900 mg, placebo | 16.8 | –6.16 vs –4.2* | 55% vs 31% | 32% vs 16% | –67.51 vs –37.10 | 47% vs 40% | ||||||
| NCT02629861 | 875 | Phase 3 (HALO) | EM | SC M: 225 mg | 12 weeks | 9.1 | –3.7 vs –2.2* | 47.7% vs 27.9% | 66.2% vs 58.4% | |||
| SC Q: 675 mg + placebo | –3.4 vs –2.2* | 44.4% vs 27.9% | NA | NA | NA | 66.3% vs 58.4% | ||||||
| NCT02621931 | 1130 | Phase 3 (HALO) | CM | SC M: 225 mg × 3 | 12 weeks | –4.6 vs –2.5* | 38% vs 18.1% | 71% vs 64% | ||||
| SC Q: 675 mg, placebo | 16.2 | –4.3 vs –2.5* | 41% vs 18.1% | NA | NA | NA | 70% vs 64% | |||||
| Galcanezumab33,34,36,70–72(LY2951742) | ||||||||||||
| NCT01625988 | 218 | Phase 2 | EM | SC every 2 weeks: 150 mg, placebo | 12 weeks | 6.85 | –4.2 vs –3.0* | 70% vs 45% | 49% vs 27% | 32% vs 17% | NA | 72% vs 67% |
| NCT02163993 | 410 | Phase 2b | EM | SC M: 5, 50, 120, 300 mg, placebo | 12 weeks | 6.65 | –3.8 vs –3.7 | NA | NA | NA | NA | 60.3% vs 51.1% |
| –4 vs –3.7 | NA | NA | 45.6% vs 51.1% | |||||||||
| –4.8 vs –3.7* | NA | NA | 51.4% vs 51.1% | |||||||||
| –4.2 vs –3.7* | 75.8% vs 61.9% | 35.5% vs 23% | 47.8% vs 51.1% | |||||||||
| NCT02614183 | 858 | Phase 3 (EVOLVE-1) | EM | SC M: 120, 240 mg, placebo | 6 months | 9.1 | –4.7 vs 2.8* | 62.3% vs 38.6% | 38.8% vs 19.3% | 15.6% vs 6.2% | NA | 65.5% vs 60.4% |
| –4.6 vs 2.8* | 60.9% vs 38.6% | 38.5% vs 19.3% | 14.6% vs 6.2% | 67.7% vs 60.4% | ||||||||
| NCT02614196 | 915 | Phase 3 (EVOLVE-2) | EM | SC M: 120, 240 mg, placebo | 6 months | 9.1 | –4.3 vs –2.3* | 59.3% vs 36.0% | 33.5% vs 17.8% | 11.5% vs 5.7% | NA | NA |
| –4.2 vs –2.3* | 56.5% vs 36% | 34.3% vs 17.8% | 13.8% vs 5.7% | |||||||||
| NCT02614261 | 1113 | Phase 3 (REGAIN) | CM | SC M: 120, 240 mg, placebo | 3 months | 19.4 | –4.8 vs –2.7* | 27.5% vs 15.4% | 8.8% vs 4.5% | 1.3% vs 0.5% | 36.2 vs 13.4 | 58% vs 50% |
| –4.6 vs –2.7* | 31.5 vs 13.4 | 57% vs 50% | ||||||||||
| Erenumab30,31,38,39,73,74(AMG334) | ||||||||||||
| NCT01952574 | 483 | Phase 2 | EM | SC M: 7, 21, 70 mg, placebo | 12 weeks | 8.7 | –2.2 vs –2.3 | 29% vs 30% | NA | NA | –19.3 vs –22.8 | 50% vs 54% |
| EM | –2.4 vs –2.3 | 34% vs 30% | –17.8 vs –22.8 | 51% vs 54% | ||||||||
| –3.4 vs 2.3* | 46% vs 30% | –34.1 vs –22.8 | 54% vs 54% | |||||||||
| NCT02066415 | 667 | Phase 2 | CM | SC M: 70, 140 mg, placebo | 12 weeks | 17.9 | –6.6 vs –4.2* | 40% vs 23% | NA | NA | –64.8 vs –55.2 | 44% vs 39% |
| CM | –6.6 vs –4.2* | 41% vs 23% | –74.5 vs –55.2 | 47% vs 39% | ||||||||
| NCT02483585 | 577 | Phase 3 (ARISE) | EM | SC M: 70 mg, placebo | 12 weeks | 8.3 | –2.9 vs –1.8* | 39.7% vs 29.5% | NA | NA | NA | 48.1% vs 54.7% |
| NCT02456740 | 955 | Phase 3 (STRIVE) | EM | SC M: 70, 140 mg, placebo | 6 months | 8.3 | –3.2 vs –1.8* | 43.3% vs 27% | NA | NA | NA | 57.3% vs 63% |
| EM | –3.7 vs –1.8 | 50% vs 27% | 55.5% vs 63% | |||||||||
| (P < 0.001) | ||||||||||||
| NCT03096834 | 246 | Phase 3b (LIBERTY) | EM | SC M: 140 mg, placebo | 12 weeks | 9.3 | –1.8 vs –10.2* | 30.3% vs 13.7%* | 12% vs 4% | 6% vs 0% | NA | 55% vs 54% |
| EMc | ||||||||||||
CGRP: calcitonin gene–related peptide; CM: chronic migraine; EM: episodic migraine; IC: inclusion criteria; IV: intravenous; M: monthly; mAb: monoclonal antibody; MD: migraine days per month; MH: migraine hours per month; N: sample size; NA: not available; Q: quarterly; SC: subcutaneous.
Phase 3 trials of the different monoclonal antibodies follow a similar design: multicentre double-blind randomised controlled trials with 3 arms, with different dosages of the drug in 2 of these. The main outcome variable is the number of migraine days per month, with patients being considered responders if this number decreases by at least 50% after 12 weeks of treatment. The mean number of migraine days/month at baseline ranged between 8.3 and 9.1 in the different trials. All monoclonal antibodies were found to be more efficacious than placebo for migraine prevention. The results of the completed phase 3 trials are summarised below.
ErenumabSTRIVE trial (NCT02456740). The study included 955 patients, assigned to 24 weeks’ treatment in 3 arms: 70 mg/month erenumab (n = 317), 140 mg/month erenumab (n = 319), or placebo (n = 319). Both dosages of erenumab were superior to placebo in reducing the number of migraine days and in the following secondary outcome measures: number of days with use of acute medication for migraine, disability scales, and daily living activity scales.30 The trial did not exclude patients with oral preventive treatments in monotherapy at a stable dosage.
ARISE trial (NCT02483585). The trial included 577 patients, who received 70 mg/month erenumab (n = 282) or placebo (n = 288) for 12 weeks. Erenumab performed better than placebo in reducing migraine days per month and in secondary outcome variables (the same as those used in the STRIVE trial).31
FremanezumabNCT02629861 trial. The trial included 875 patients assigned to 3 arms: 225 mg fremanezumab monthly (n = 290), 675 mg fremanezumab quarterly (n = 291), or placebo (n = 294). Fremanezumab performed significantly better than placebo in reducing the number of migraine days per month and the number of days with use of acute medication for migraine, as well as improving scores on the Migraine Disability Assessment (MIDAS).32 The trial did not exclude patients taking oral preventive treatments in monotherapy at a stable dosage.
GalcanezumabEVOLVE1 (NCT02614183) and EVOLVE2 trials (NCT02614196). These trials included 858 and 915 patients, respectively, who were assigned to 6 months’ treatment in 3 arms: galcanezumab 120 mg, galcanezumab 240 mg, or placebo. Both dosages were superior to placebo in the primary outcome variable and in quality of life scales.33,34
EptinezumabPROMISE1 trial (NCT02974153). The trial included 888 patients assigned to 4 arms: 300 mg, 100 mg, or 30 mg eptinezumab; or placebo. All 3 doses of the drug performed significantly better than placebo in reducing the number of migraine days per month and in the number of patients achieving a ≥ 50% reduction in the frequency of attacks.35
Efficacy in episodic migraineCurrently, results from phase 3 trials are only available for fremanezumab and galcanezumab. Data from the PROMISE2 trial (NCT02559895) on eptinezumab have not been published. Phase 2 trials have demonstrated the efficacy of erenumab, and open-label follow-up studies of the drug are underway; no phase 3 trials have been conducted with patients with CM.
REGAIN trial (NCT02614261). This trial of galcanezumab demonstrated a mean reduction of 4.3 migraine days per month in patients receiving 120 mg of the drug (n = 226) and 4.6 days in those receiving 240 mg (n = 220), compared to placebo.36
NCT02621931 trial. The study included 1130 patients, assigned to 16 weeks’ treatment in 3 arms: 675 mg fremanezumab (n = 376), 225 mg fremanezumab (n = 379), or placebo (n = 375). Fremanezumab achieved a greater reduction in the number of headache days per month, and a better response in the secondary outcome variables (migraine days per month and Headache Impact Test [HIT-6] score).37 The trial did not exclude patients taking oral preventive treatments in monotherapy at a stable dosage.
Efficacy in refractory migraineDifferent trials have evaluated treatment with fremanezumab (FOCUS trial), erenumab (LIBERTY trial), and galcanezumab (CONQUER trial) in patients presenting lack of response to 2-4 classes of treatment. So far, results have been published for erenumab in patients with EM (LIBERTY trial; NCT03096834): erenumab 140 mg (n = 121) achieved a 50% reduction in migraine days per month in 30% of patients, as compared to 14% in the placebo group (n = 125).38 Results are also available from a sub-analysis of erenumab in patients with CM and previous failure of one or 2 treatments.39
Safety dataUnlike gepants, monoclonal antibodies are not metabolised in the liver; furthermore, as they do not cross the BBB, no adverse reactions are expected at the level of the central nervous system, with the exception of such structures as the pituitary gland, which are not protected by the BBB. However, it has been suggested that prolonged antagonism of CGRP may represent a risk in the different organs and systems in which the neuropeptide plays a key role in animal studies: the skin (scarring), gastrointestinal system (mucosal protection, motility), cardiovascular system (protection against cardiac and brain ischaemia and hypertension), bone metabolism, and glomerular filtration.
To date, very similar rates of adverse events have been reported in the active and placebo groups, and no severe adverse events have been detected; however, the duration of the trials is relatively short for a chronic condition such as migraine. Noteworthy reactions include pain at the injection site for all drugs, with little or no difference with respect to placebo, induration or erythema with fremanezumab, and pruritis or local reactions with galcanezumab. No significant differences are reported with respect to placebo in the frequency of respiratory or urinary infection, fatigue, or nausea, and no vascular events have been recorded. Drop-out rates were low in all trials, and drop-outs due to adverse events were very infrequent (always < 4%), unlike the figures recorded for oral preventive treatments.40 However, as adverse events cannot be ruled out in the long term, there is a need for registries to gather additional safety data.
What are the benefits of monoclonal antibodies over current treatments for episodic and chronic migraine?Compared to the other options currently available for migraine prevention, monoclonal antibodies represent a significant advance, as all the therapeutic tools used previously (eg, antihypertensives, antidepressants, neuromodulators) were originally developed in other branches of medicine. The clinical development of the new monoclonal antibodies was similar to that of antibodies used to treat other diseases, and current evidence indicates potential benefits for both EM and CM.
All patients with CM and up to 40% of those with EM are estimated to be eligible for preventive treatment to reduce the impact of the disease and the resulting disability; however, only a very small percentage ultimately receive the drugs.41 The currently available treatments present several issues in terms of efficacy, tolerability, treatment adherence, and drug interactions. Treatment is suspended in approximately one in 5 patients due to adverse events,42 with only one in 5 showing proper treatment adherence at one year.43 In the United States, periodic OnabotA infiltration is currently the only treatment specifically approved for CM, the most disabling form of the disease.44
In the light of the above, the new monoclonal antibodies present the following differences with respect to classical treatments:
- 1
Route and schedule of administration: with the exception of OnabotA and anaesthetic nerve block, most of the available preventive treatments are administered orally on a daily basis. However, monoclonal antibodies are administered subcutaneously (in the deltoid, thigh, or abdomen) on a monthly or quarterly basis, with the exception of eptinezumab, which is administered intravenously every 3 months. An important difference is that this type of molecule will probably be dispensed at hospital pharmacies, like other monoclonal antibodies; therefore, healthcare centres will have to implement internal administrative procedures for the administration of these drugs. As a result, we may expect that, at least initially, these treatments will be reserved for refractory cases and may be subject to some kind of specific evaluation at specialised headache clinics.
- 2
Action mechanism: while other migraine treatments (eg, triptans, gepants, methysergide, and even OnabotA) act on the CGRP pathway, no previous treatment has so selectively blocked this molecule.
- 3
Adherence: one of the main problems frequently reducing the efficacy of the current treatments is poor adherence on the part of patients and incorrect compliance with treatment schedules issued by clinicians. We may expect to see greater adherence to these new treatments as they will be administered at the hospital itself or with autoinjectors that require strict patient education.
- 4
Efficacy and safety: this point is addressed in detail elsewhere in the article. It is too early to determine what aspects of efficacy may show improvements over classical treatments, given the current lack of comparative studies.45 It should be noted that, unlike with oral preventive treatments, response to these treatments occurs early, in the first days after treatment onset. In terms of safety, the data available from different clinical trials are very promising, both due to the absence of severe adverse events and because of the low rates of adverse events in general; we may expect this safety profile to improve treatment adherence. Adverse reactions are a frequent problem with the current treatments.
- 5
Interactions and contraindications: to date, no relevant interactions with monoclonal antibodies have been reported. This represents a significant advantage: given the comorbidities and medication overuse associated with migraine, oral preventive treatments can present interactions with one another or with other groups of drugs.46 To date, no contraindications have been defined; these drugs should be avoided during pregnancy.
- 6
Simplicity of treatment schedule: in the published trials, the treatment was administered in monotherapy, with clear evidence of efficacy. Given that the great majority of patients, and especially those with more refractory migraine, receive complex treatments, it is highly likely that treatment schedules will be simplified in many cases. Furthermore, no dose titration is needed for monoclonal antibodies. It remains to be determined how monoclonal antibodies will be used for adjunctive therapy or in polytherapy schedules, and what drug combinations may be the most rational.
It is difficult to characterise the profile of patients who respond to monoclonal antibodies targeting the CGRP receptor or ligand. The clinical trials conducted to date have found these drugs to be efficacious in diverse contexts: migraine with and without aura, episodic and chronic migraine, migraine responsive and refractory (and even naïve) to previous preventive treatments, migraine with and without symptomatic medication overuse, and migraine with and without associated comorbidities. As a result of these data, the United States Food and Drug Administration has already approved erenumab, fremanezumab, and galcanezumab for the preventive treatment of migraine, with no restriction or limitation. In theory, any patient presenting 4 or more migraine attacks per month would be eligible to receive these treatments.
Despite this, and in the light of the available data, we must acknowledge that not all patients benefit equally. Evidence from the published trials indicates the existence of a group of “hyper-responders,” a group presenting a moderate response, and another group of patients with little or no response.47 The problem is that no predictive factors of treatment response have yet been identified, and we may only hypothesise about which patients may respond best to the treatment.
Given the characteristics of monoclonal antibodies, it seems reasonable that the main candidates for this treatment, in whom we would expect to achieve the greatest benefits, would be patients with high frequency and intensity of migraine attacks, presenting recurrent, sustained release of CGRP. Scheduled determination of plasma CGRP levels during and between migraine attacks supports this hypothesis.20 To corroborate this assertion, it would be highly beneficial to demonstrate that patients with migraine presenting high plasma CGRP levels respond better to monoclonal antibodies and that levels of the peptide decrease after administration of the antibodies in patients with a better response. It remains to be determined whether this hypothesis can be evaluated for erenumab (the only monoclonal antibody targeting the CGRP receptor, rather than the ligand), as plasma CGRP determination may not be as valuable for this drug as for other monoclonal antibodies. In theory, patients with greater CGRP pathway activity should respond the best to monoclonal antibodies. On the contrary, patients in whom other pathways play a greater role (eg, VIP, PACAP 38, or other inflammatory neuropeptides related to migraine attacks and their perpetuation or migraine transformation) should present a clearly inferior response.
When is migraine considered to be treatment-resistant?Refractory migraine inherently involves greater disability and a greater impact on quality of life. Although its prevalence in the general population is unknown, refractory migraine accounted for 5.1% of all patients attended at a specialised headache clinic in the study by Irimia et al.48 No consensus diagnostic criteria have been established49–56 and, despite recommendations by expert groups, it is not included in the third edition of the International Classification of Headache Disorders.57
In clinical practice, the term is used to describe patients with migraine associated with significant disability and impact on quality of life and lack of response to several preventive treatments administered either in monotherapy or in combination therapy.
In 2008, the Refractory Headache Special Interest Section of the American Headache Society proposed a series of diagnostic criteria for refractory migraine, which are valid both for episodic and for chronic forms.50 Headache must cause interference with quality of life despite modification or optimisation of the 3 basic pillars of therapeutic management of migraine: triggers and lifestyle factors, acute symptomatic treatment, and preventive treatment. At least 2 of 4 treatment groups (beta-blockers, anticonvulsants, calcium channel blockers, and tricyclic antidepressants), either in monotherapy or in combination, must have resulted in treatment failure. Non-responsiveness is defined as lack of response to treatment of at least 2 months’ duration at the optimal dose (in a subsequent survey of members of the American Headache Society, approximately 40% considered it appropriate to increase the number of preventive treatments to 3 or even 4 drugs).54
The authors also require failure of at least 2 possible strategies in the symptomatic treatment of attacks: 1) both a triptan and dihydroergotamine in an intranasal or injectable formulation; or 2) either a non-steroidal anti-inflammatory drug or combination analgesic therapy. Furthermore, assessing secondary disability (MIDAS score ≥ 11) as an operational criterion introduces a new concept, the impact of migraine on quality of life. Finally, these criteria permitted the possible co-presence of medication overuse.
In 2014, the European Headache Federation published its diagnostic criteria for refractory migraine, which only apply to CM.56 Diagnosis is based on the failure of at least 3 groups of treatments (including OnabotA), which must be administered for at least 3 months. The criteria also establish the need to rule out medication overuse headache through withdrawal of the drug, either through pharmacological (oral/intravenous) or patient education measures.
In our opinion, it is difficult to opt for one definition over the other. The American criteria have the advantage that they consider disability and the response to symptomatic treatment (although intranasal dihydroergotamine is not available in Europe) and do not require withdrawal of any overused medication.50 Furthermore, they are applicable to EM; the boundary between chronic and episodic migraine is frequently permeable, as has been demonstrated in epidemiology studies.58 The European definition more clearly establishes the procedure for ruling out secondary headache and accounts for preventive treatment with OnabotA.56
Based on critical analysis of both definitions, we suggest the possibility of OnabotA treatment of EM, in line with the GECSEN guidelines, if preventive treatments fail or are poorly tolerated.59,60 Furthermore, it would be reasonable to include anaesthetic block of the greater occipital nerve as a possible therapeutic option in definitions of refractory EM and refractory CM (level of evidence IV and IIIb, respectively).61 It also seems inappropriate that the drugs with the greatest level of evidence in CM (topiramate and OnabotA) should be given the same weight as those considered second-line treatments in the management of the condition.56
In terms of pharmacoeconomics, what do anti-CGRP monoclonal antibodies contribute to the treatment of migraine?At the time of writing, no data are available on the pricing and reimbursement of monoclonal antibodies in Spain; the commercialisation of these drugs represents a considerable advance in the preventive treatment of migraine.62 Once the efficacy and safety of these treatments are demonstrated, clinicians must be provided with complementary information on their pharmacoeconomics. This consideration is highly important given the significant prevalence of migraine, the limited economic resources of the healthcare system, and the current commercial availability of other efficacious treatments. This type of study enables us to establish which treatment is the most cost-effective. Economic evaluation of preventive drugs for migraine must take into account their impact on direct costs (medical consultations, emergency department visits, cost of the drug), indirect costs (loss of productivity), and the patient’s quality of life.
Currently, very little information is available on the pharmacoeconomics of monoclonal antibodies, and no studies have compared their efficacy and costs against those of oral preventive treatments or OnabotA. Recent publications have shown that erenumab reduces the direct and indirect costs associated with migraine in patients with EM and CM (who had previously used at least one preventive treatment), and increases the number of quality-adjusted life years, in comparison to no preventive treatment.63 Cost calculations are based on the American healthcare system, and it is important to stress that the actual results could be even better than those reported, as the study was performed before erenumab was commercialised, resulting in cost estimates higher than the drug’s final market price.63
The American Institute for Clinical and Economic Review, an independent, non-profit research body, recently published its final evidence report on the effectiveness of monoclonal antibodies for migraine and whether their price was commensurate with their clinical benefits (long-term cost-effectiveness).64 The long-term value of erenumab (the only antibody for which a public price was available) was considered to be intermediate in adults with CM and intermediate-low for EM.
This subject will probably be addressed in new publications in the coming months, as research on the pharmacoeconomics of new medications is a prerequisite for pricing and reimbursement negotiations in different European countries. Furthermore, the British National Institute for Health and Care Excellence guidelines are expected to be published in late 2018; the document will analyse the clinical effectiveness and cost-effectiveness of different CGRP monoclonal antibodies, as part of the United Kingdom’s procedure for the commercialisation of the drugs.
Monoclonal antibodies are safe, effective, and well tolerated.65 It is reasonable to expect that the reduction in the number of migraine days and in disability will be associated with a decrease in the direct and indirect costs of the condition and an improvement in patients’ quality of life. The limited available evidence supports this hypothesis. However, there is a need for further pharmacoeconomic studies to provide additional information on the potential benefits of these treatments and to help clinicians to make rational decisions in choosing between the current treatment alternatives; this will result in better use of the available resources and an increase in care quality.
FundingThis study has received no funding of any kind.
Conflicts of interestThe authors have no conflicts of interest to declare.
Dr Belvís has received honoraria from Allergan, Chiesi, Novartis, Teva, Juste, and Eli Lilly.
Dr Guerrero Peral has received honoraria from Allergan, Alder, Amgen, Chiesi, Eli Lilly, Novartis, and Teva.
Dr Díaz Insa has received honoraria from Allergan, Alter, Kern Pharma, Lundbeck, Novartis, and Teva.
Dr Gago Veiga has received honoraria from Allergan, Eisai, Novartis, Teva, and UCB Pharma.
Dr Cuadrado has received honoraria from Almirall, Allergan, Juste, and Novartis.
Dr Huerta has received honoraria from Almirall, Allergan, Chiesi, Eisei, Kern Pharma, MSD, Eli Lilly, Teva, and Zambón.
Dr Irimia has received honoraria from Alder, Allergan, Eli Lilly, and Novartis.
Dr Láinez has received honoraria from Allergan, Amgen, ATI, Bial, Boehringher, Chiesi, Eisai, ElectroCore, Eli Lilly, Medtronic, Novartis, Otsuka, Roche, Teva, and UCB.
Dr Latorre has received honoraria from Allergan, Chiesi, Novartis, and Teva.
Dr Leira has received fees for clinical trials, lectures, and consulting from Allergan, Amgen, Chiesi, Eli Lilly, Novartis, and Teva.
Dr Pascual has received honoraria from Almirall, Amgen, Novartis, Stendhal, and Teva.
Dr Porta-Etessam has received honoraria from Allergan, Chiesi, Eisai, Exeltis, Grunenthal, Novartis, and Teva.
Dr Pozo-Rosich has received honoraria from Allergan, Almirall, Amgen, Chiesi, Eli Lilly, Novartis, and Teva.
Dr Sánchez del Río has received honoraria from Allergan, Chiesi, Eli Lilly, Novartis, and Teva.
Dr Santos Lasaosa has received honoraria from Allergan, Amgen, Chiesi, Eisai, Exeltis, Novartis, and Teva.
Dr Viguera has received honoraria from Allergan and Novartis.
Please cite this article as: Santos-Lasaosa S, Belvís R, Cuadrado ML, Díaz-Insa S, Gago-Veiga A, Guerrero-Peral AL, et al. CGRP en migraña: de la fisiopatología a la terapéutica. Neurología. 2022;37:390–402.
