



Chronic hypoperfusion in rats produces memory and learning impairments due to permanent occlusion of commun carotid arteries (POCCA). Molecular mechanisms leading to behavioural disorders have been poorly studied. For this reason, the aim of the present study was to characterise oxidative metabolism disorders and their implications in memory and learning impairments.
MethodsSuperoxide dismutase (SOD) and catalase (CAT) activities were determined in cortex, hippocampus and striatum homogenates at 24hours and at 22 days after the lesion. Haematoxylin–eosin staining and glial fibrillary acidic protein (GFAP) immunoreactivity were performed on coronal sections. Behavioural impairments were explored using the Morris water maze (MWM). Escape latencies were determined in all behavioural studies.
ResultsThe lesion induced a significant increase (P<.01) in CAT activity in the cortex at 24hours, while SOD activity was significantly higher (P<.01) in the cortex and hippocampus at 22 days. An intense vacuolization was observed in the cortex and striatum as a result of the lesion. A neuronal loss in the striatum and hippocampus was observed. The glial reaction increased in the cortex and striatum. Visual alterations were observed in the lesion group with the lowest evolution time (P<.001). Escape latencies, corresponding to MWM schemes for long-term and short-term memory evaluation increased significantly (P<.05) in both groups of lesioned animals.
ConclusionIt was concluded that changes in SOD and CAT activities indicate a possible implication of oxidative imbalance in the pathology associated with chronic cerebral hypoperfusion. In addition, the POCCA model in rats is useful for understanding mechanisms by which cerebral hypoperfusion produces memory and learning impairments.
La hipoperfusión cerebral en ratas, mediante la oclusión permanente de las arterias carótidas comunes (OPACC), induce alteraciones de la memoria y el aprendizaje. Los mecanismos moleculares han sido poco estudiados y el objetivo del trabajo consiste en caracterizar las alteraciones del metabolismo oxidativo, de la memoria y del aprendizaje.
MétodosMediante la OPACC se determinó a las 24 h y 22 días de la lesión la actividad de la superóxido dismutasa y la catalasa en el hipocampo, la corteza y el cuerpo estriado. Se realizó una tinción con hematoxilina-eosina y un marcaje con GFAP de cortes coronales. Los trastornos conductuales se exploraron mediante la prueba del laberinto acuático de Morris.
ResultadosLa lesión indujo un incremento (p<0,01) de la actividad de la catalasa en la corteza a las 24 h, mientras que la superóxido dismutasa aumentó significativamente (p<0,01) en la corteza y el hipocampo a los 22 días. Se observó una intensa vacuolización y pérdida neuronal. La respuesta glial estuvo incrementada en la corteza y el cuerpo estriado. Fue perceptible la afectación en la visión (p<0,001), y las latencias de escape al evaluar la memoria a largo y corto plazo aumentaron considerablemente (p<0,05) en ambos grupos de animales lesionados.
ConclusionesLos cambios en las actividades de las enzimas apuntan a una posible implicación del disbalance oxidativo en la patología asociada a la hipoperfusión cerebral crónica. La OPACC resulta útil para entender los mecanismos por los cuales la hipoperfusión cerebral conduce a las alteraciones de los procesos de memoria y aprendizaje.
It is a well-known fact that while stroke incidence increases with age, strokes are not necessarily fatal.1 Ischaemic events occur due to occlusion of a major artery that causes a decrease in blood flow.2
The brain possesses specific traits which make its tissue highly vulnerable to the effects of oxidative stress.3,4 An ischaemic event will result in a marked increase in reactive oxygen species (ROS). However, enzymatic antioxidant defence systems found in the brain neutralise such highly reactive species. This defence system relies on cooperative action between intracellular enzymes, which include superoxide dismutase (SOD) and catalase (CAT).5
Ischaemia has historically been considered untreatable, and there are no effective treatment options even today.6 One facet of the search for new treatment therapies focusing on neuroprotection or neurorestoration of the damaged tissue is the development of experimental animal models.7 The model for permanent occlusion of the common carotid arteries (POCCA) in rats reproduces both the ischaemic event in its early stages and the subsequent oligaemia once cerebral hypoperfusion has become chronic.7 The hippocampus is part of the brain that has received the most attention from studies on POCCA-induced neuropathological alterations. Cerebral hypoperfusion has been associated with impaired memory and learning; both of these processes involve the hippocampus.7,8 It has been suggested that, in addition to the hippocampus, the striatum and the cortex are also highly susceptible to ischaemic events.5
The purpose of this study is to describe alterations in oxidative metabolism in the cortex, striatum, and hippocampus, and the effect of these changes on memory and learning disorders in a rat model of cerebral hypoperfusion.
Materials and methodsExperimental subjectsSubjects were male Sprague-Dawley rats from the National Center for the Production of Laboratory Animals (Havana, Cuba). Rats were kept in standard laboratory conditions at a mean temperature of 22°C±2°C with a 12/12hour light–dark cycle and free access to food and water. The established ethical principles for animal research were followed throughout all experiments.9 Rats had a mean weight of 311g±4.37g at the time the lesion was induced.
Surgical procedureThe 42 rats in the model group were anaesthetised with an intraperitoneal injection of 350mg chloral hydrate per kilogram body weight. The rats’ necks were incised and common carotid arteries were permanently ligated using suture silk 3-0. Rats in the control group (n=30) were subjected to the same surgical procedure but did not undergo POCCA.
Study of oxidative metabolismObtaining biological samplesFollowing the surgical procedure, at 24hours (10 experimental, 9 control) and at 22 days (10 experimental, 5 control), the animals were deeply sedated with an intraperitoneal injection of chloral hydrate (700mg/kg of body weight) and decapitated. Brains were removed so as to dissect areas of the brain (cortex, hippocampus, and striatum) from both hemispheres. Tissues were preserved at −70°C until they were used.
Tissue homogenisationAreas of the brain were homogenised using a solution of Tris 1mol/L sucrose 0.25mol/L with a pH of 7.4 in a homogeniser at 1000rpm in an ice bath. The product was placed in the centrifuge during 15minutes at 4°C and 14000rpm; the supernatant was preserved frozen at −70°C until it was processed.
Determining superoxide dismutase activityWe used the Marklund method10 based on the ability of SOD to inhibit the pyrogallol reaction. Delipidation of the samples was performed using an aliquot of 100μL of homogenate and adding 30μL chloroform and 50μL methanol. The mixture was shaken for 1minute in a laboratory shaker and centrifuged at 4°C and 3000rpm for 20minutes, after which the supernatant was collected. The buffer was Tris 50mmol/L–HCl 20mmol/L–EDTA 10mmol/L with a pH of 8.2. We added 540μL of the buffer, 30μL of the sample, and 30μL of pyrogallol. The mixture was shaken and the optical density (OD) value was obtained from duplicate measurements and recorded as 420nm at 70seconds. One unit of enzyme activity was defined as the amount of enzyme necessary to achieve 50% inhibition of pyrogallol auto-oxidation.
Determining the enzymatic activity of catalaseCAT activity was determined by using Aebi's spectrophotometric method11 to observe the decomposition of H2O2. Our trial used a phosphate buffer (KH2PO4 0.12mol/L–K2HPO4 0.12mol/L) with a pH of 7.4 and a 13mmol/L H2O2 substrate solution in a phosphate buffer. During the trial, 375μL of phosphate buffer, 204μL of substrate solution, and 21μL of homogenate were mixed and shaken. OD values at 240nm were measured every 2seconds during 1minute in a variable temperature cell set to 37°C. OD values were obtained from duplicate measurements. One unit of enzyme activity was defined as the amount of enzyme necessary in order to transform 1μmol of H2O2 in 1minute at 37°C.
Measuring total proteinsOne hundred microlitres each of water (blank sample), a reference sample, and our sample were then mixed with 2.0mL Bradford reagent (0.1mg/mL Coomassie blue in 8.5% H3PO4 and 4.75% ethanol). OD was measured at 595nm. Calibration was performed using bovine serum albumin at its extinction coefficient of 280nm (k=0.68mLmg−1).
Evaluation of cell damageFor purposes of the histological study, animals with POCCA (n=3) and controls with mock lesions (n=3) were anaesthetised with chloral hydrate 7 days after cerebral hypoperfusion surgery. They received a perfusion of 250mL 0.9% sodium chloride solution and 250mL of a 10% formalin flush. Rat brains were placed in an automatic tissue processor (Histokinette). Brains were dehydrated in a series of graded alcohol baths, the alcohol was removed with xylene, and the tissue was subjected to paraffin infiltration. Coronal slices 6μm thick were stained with haematoxylin and eosin stain and alternately subjected to the procedure for immunohistochemical detection of glial fibrillary acidic protein (GFAP). Slices were treated with 20% foetal bovine serum/Triton X-100 0.25% in a saline phosphate buffer during 30minutes and incubated overnight at 4°C with polyclonal anti-GFAP antiserum (1:500), followed by incubation with biotin anti-mouse IgG1 antibody (1:500) for 1hour at room temperature. Slices were submerged in a streptavidin–biotin-peroxidase conjugate (1:100) for 1hour and the reaction was observed with 0.02% H2O2 and 0.05% diaminobenzidine as a chromogen. The reaction was terminated by adding regular water. Slices were dehydrated with a series of graded alcohol baths, rinsed in xylene, and observed under an optical microscope.
Evaluation of behaviour disordersThe Morris water maze (MWM) is a procedure in which researchers measure the time it takes animals to locate a platform measuring 11cm in diameter, whether it is visible or hidden, in order to escape from a pool of water (escape latency). Each rat was given a maximum of 60seconds to search for a way out of a circular tank 1.5m in diameter and filled with water to a depth of 40cm. A camera trained over the centre of the water tank enabled data acquisition, and data were processed using SMART software version 2.0, copyright Panlab, 2001.
Behavioural tests were conducted in 2 different phases: in 1 group of animals at 18 days post-lesion (model group n=9, mock lesion group n=8) and in the other group at 33 days post-lesion (model group n=10, mock lesion group n=5).
Evaluating sensorimotor and motivational deficitsThe platform was placed so that it was visible, and each rat completed 8 trials at 22 and 37 days post-lesion. Each trial began by placing the rat at a randomly selected position along the perimeter of the tank.
Evaluating long-term or spatial reference memoryIn this test, which was conducted between days 18–21 and days 33–36 post-lesion, the rats had to find the platform, now concealed, at its previous location. Each rat had 29 trials (8 trials in the first 3 days and 5 trials on the last day).
Evaluating short-term or working memorySpatial reference memory was tested at 33–35 and at 48–50 days after injury. Animals were tested during 3 days, and the position of the hidden platform was changed every day. The 4 trials were conducted in 3 days; the time elapsed between a trial and the following one was 20seconds at first, followed by 20minutes, and finally followed by 2hours.
Statistical methodThe recorded data were processed using professional software: Statistica for Windows, version 6.0, Copyright Statsoft Inc., 1996. Values were shown as the mean±standard error of the mean. We determined whether or not the data were normally distributed using the Kolmogorov–Smirnov test. Since the Levene test did not reveal homogeneity of variance, we performed a non-parametric data analysis.
Comparisons of antioxidant enzymes between animals in the mock lesion group and those in the model group were performed using the Mann–Whitney U-test. The same test was used to compare results from behavioural tests in the control group and in the mock lesion group, and between animals in the experimental group at different points in time. The Wilcoxon rank-sum test was used to compare behavioural task performance by the same group of animals at different times.
ResultsOxidative metabolismThere were no significant differences in antioxidant enzyme activity between animals in different control groups, so all control groups were considered to be the same.
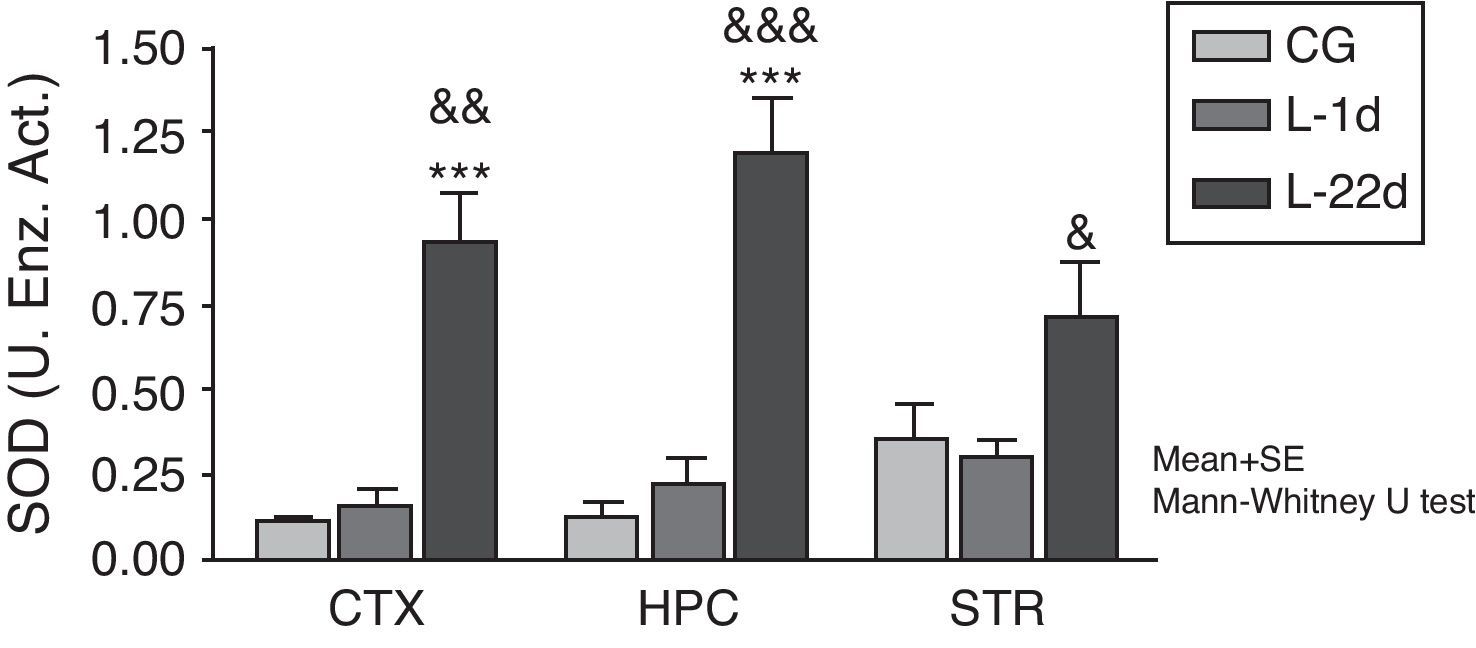
At 24hours after occlusion, there were no variations in SOD activity in any of the brain areas that were studied (Fig. 1). There was a significant increase in SOD activity in the cortex and hippocampus at 22 days post-lesion (P<.001). On the other hand, animals with cerebral hypoperfusion at 22 days post-lesion also showed a significant increase in enzymatic activity in the 3 areas of the brain that were studied (P<.05) compared to animals that were euthanised at 24hours post-lesion.
 and the model group at 24hours post-lesion (&P<.05; &&P<.01; &&&P<.001). Bars represent mean values±standard error of the mean. CTX: cortex; STR: striatum; CG: control group; HPC: hippocampus; L-1d: group at 24hours post-lesion; L-22d: group at 22 days post-lesion; SOD: superoxide dismutase.")
Activity of superoxide dismutase in the cortex, hippocampus, and striatum of rats subjected to permanent occlusion of common carotid arteries. The graph shows enzyme activity among model group animals at 22 days post-lesion compared to the control group (***P<.001) and the model group at 24hours post-lesion (&P<.05; &&P<.01; &&&P<.001). Bars represent mean values±standard error of the mean. CTX: cortex; STR: striatum; CG: control group; HPC: hippocampus; L-1d: group at 24hours post-lesion; L-22d: group at 22 days post-lesion; SOD: superoxide dismutase.
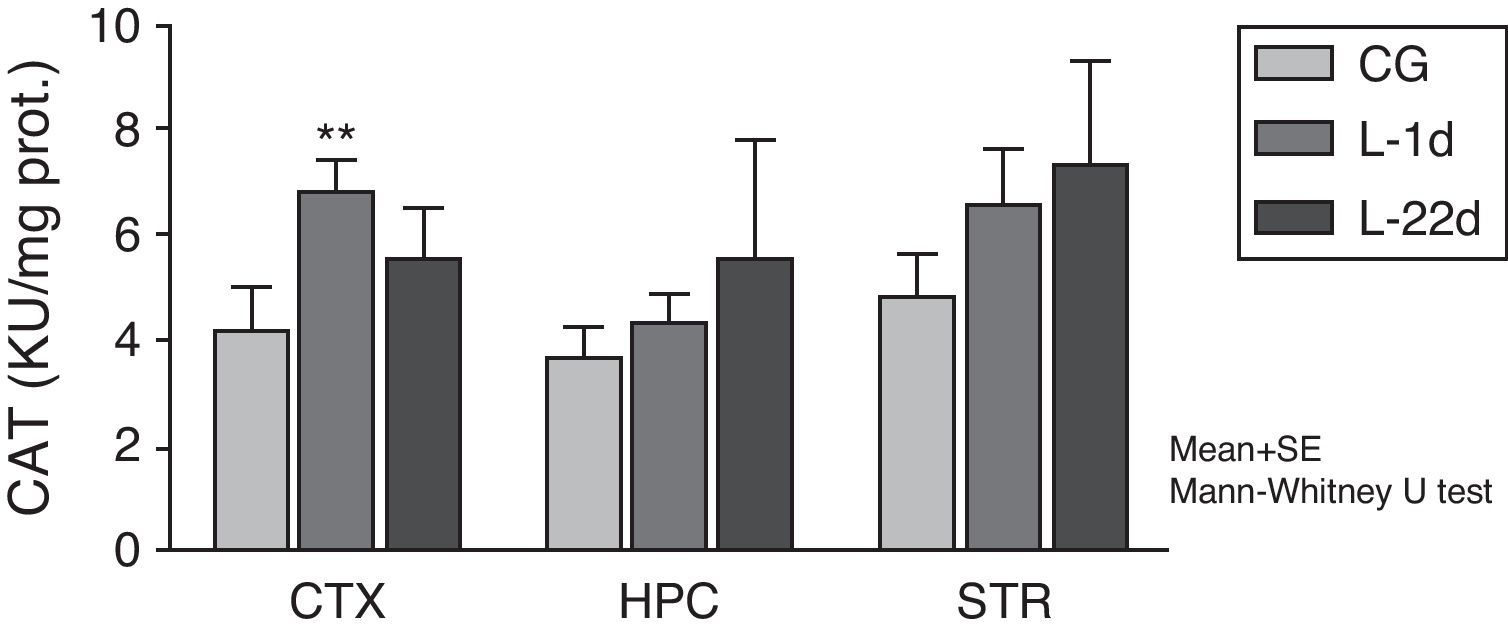
The POCCA procedure led to increased CAT activity at 24hours, but this increase was only significant in the cortex (P<.01) (Fig. 2). At 22 days after carotid occlusion, this increase was no longer statistically significant.
. Bars represent mean values±standard error of the mean. CAT: catalase; CTX: cortex; STR: striatum; CG: control group; HPC: hippocampus; L-1d: group at 24hours post-lesion; L-22d: group at 22 days post-lesion.")
Activity of catalase in the cortex, hippocampus, and striatum of rats subjected to permanent occlusion of common carotid arteries. The graph shows enzymatic activity in model group animals compared to control group animals (**P<.01). Bars represent mean values±standard error of the mean. CAT: catalase; CTX: cortex; STR: striatum; CG: control group; HPC: hippocampus; L-1d: group at 24hours post-lesion; L-22d: group at 22 days post-lesion.
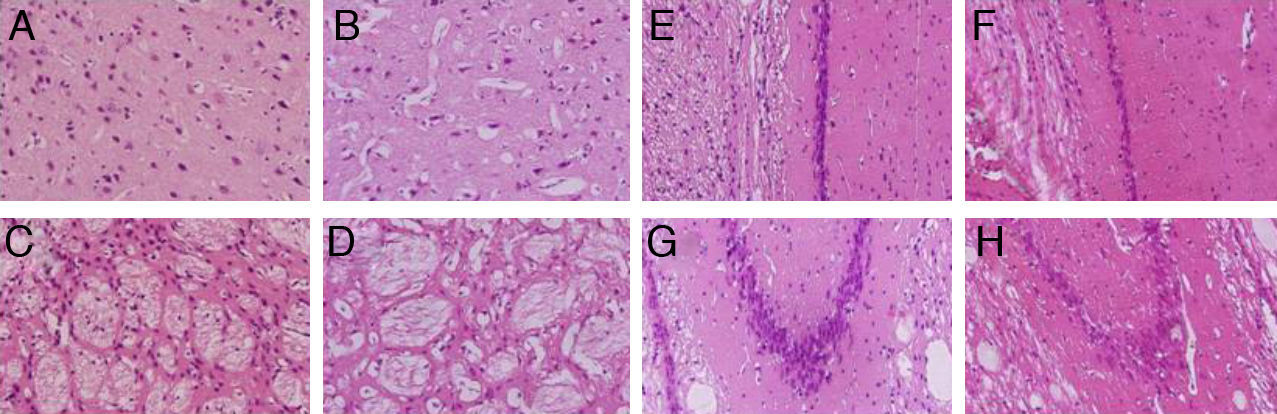
We observed pronounced vacuolisation in the cortex and striatum. However, decreases in neuronal density were only recorded in the striatum and in the CA1 and CA3 populations of the hippocampus (Fig. 3).
, the striatum (C and D), CA1 (E and F), and CA3 (G and H) at 40× magnification. Columns on the left show slices taken from control-group animals; columns on the right show sections from animals with cerebral hypoperfusion at 7 days post-lesion.")
H&E stains of coronal slices of the cortex (A and B), the striatum (C and D), CA1 (E and F), and CA3 (G and H) at 40× magnification. Columns on the left show slices taken from control-group animals; columns on the right show sections from animals with cerebral hypoperfusion at 7 days post-lesion.
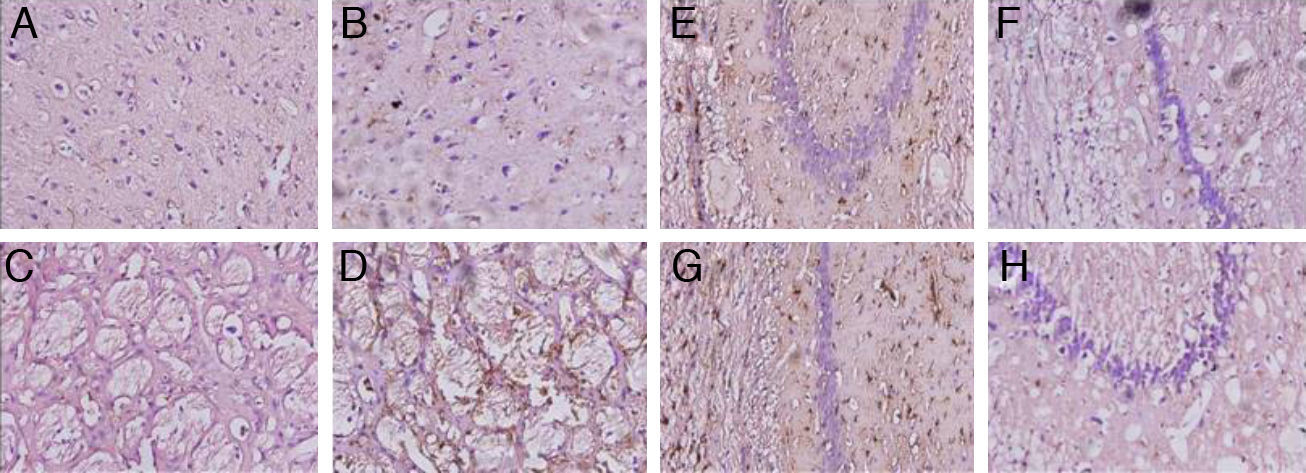
Animals with cerebral hypoperfusion displayed an increase in glial markers in the cortex and the striatum, with decreased glial response in CA1 and CA3 cells compared to control animals (Fig. 4).
 the striatum (C and D), CA1 (E and F), and CA3 (G and H) at 40× magnification. Columns on the left show slices taken from control-group animals; columns on the right show sections from animals with cerebral hypoperfusion at 7 days post-lesion.")
Microphotographs showing coronal slices from rat brains with glial fibrillary acid protein immunostaining in the cortex (A and B) the striatum (C and D), CA1 (E and F), and CA3 (G and H) at 40× magnification. Columns on the left show slices taken from control-group animals; columns on the right show sections from animals with cerebral hypoperfusion at 7 days post-lesion.
Control groups matched to model groups for different times post-lesion displayed no significant differences on any of the behavioural tests. All of these control groups were therefore considered to be a single control group.
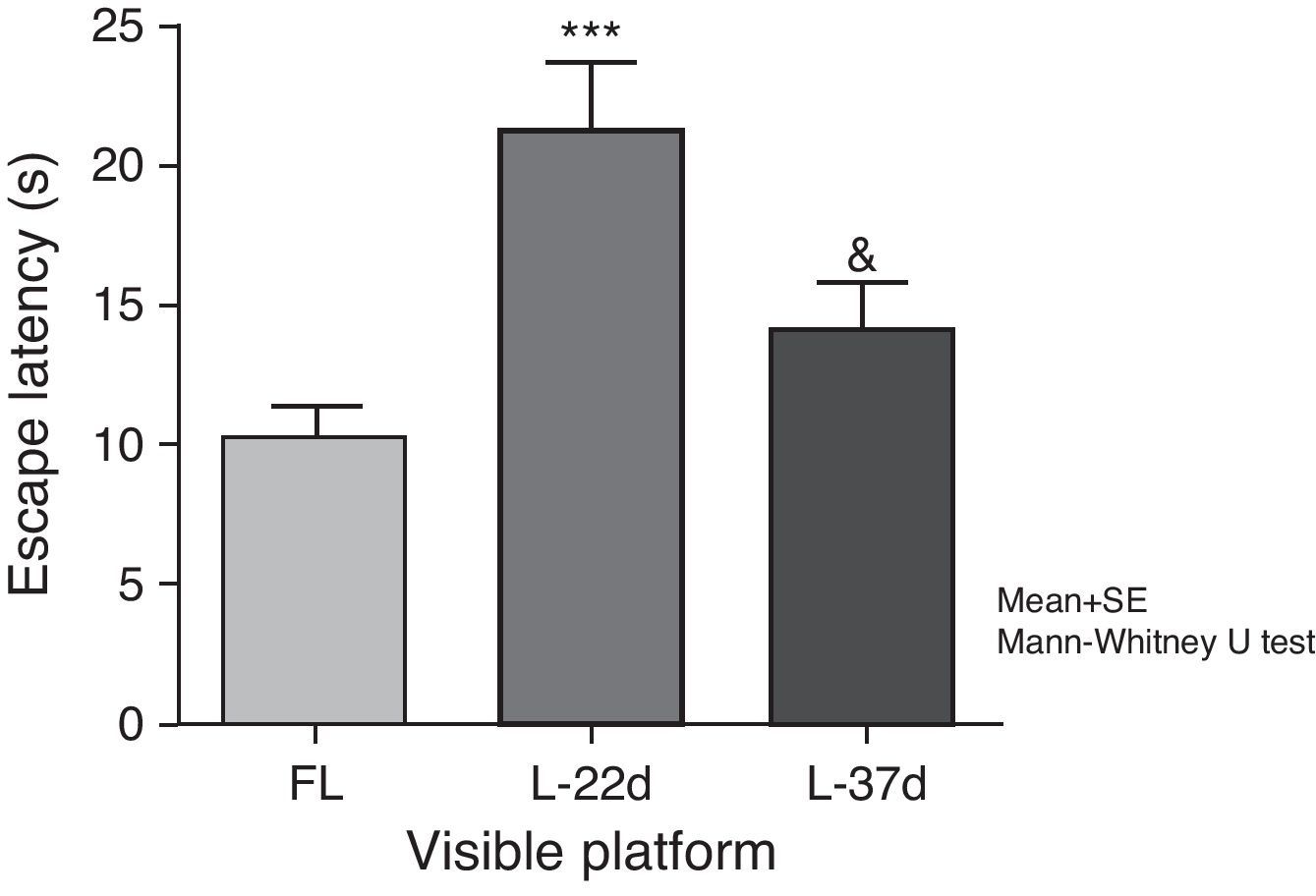
Sensorimotor and motivational deficitsAnimals with cerebral hypoperfusion assessed at 22 days post-lesion required a significantly longer time to find the escape platform than control animals did (P<.001). At 37 days after carotid occlusion, these differences were no longer statistically significant. The animals at more advanced post-lesion stages required significantly less time to find the platform than animals which had been experiencing cerebral hypoperfusion during a shorter time period (P<.05) (Fig. 5).

Influence of chronic cerebral hypoperfusion on sensorimotor and motivational deficits. For this task, the escape platform was visible. ***P<.001: escape latency in the model group at 22 days post-lesion was longer than in the control group. &P<.05: escape latency in the model group at 37 days post-lesion was shorter than in the model group at 22 days post-lesion. Bars represent mean values±standard error of the mean. CG: control group; L-22d: group at 22 days post-lesion; L-37d: group at 37 days post-lesion.
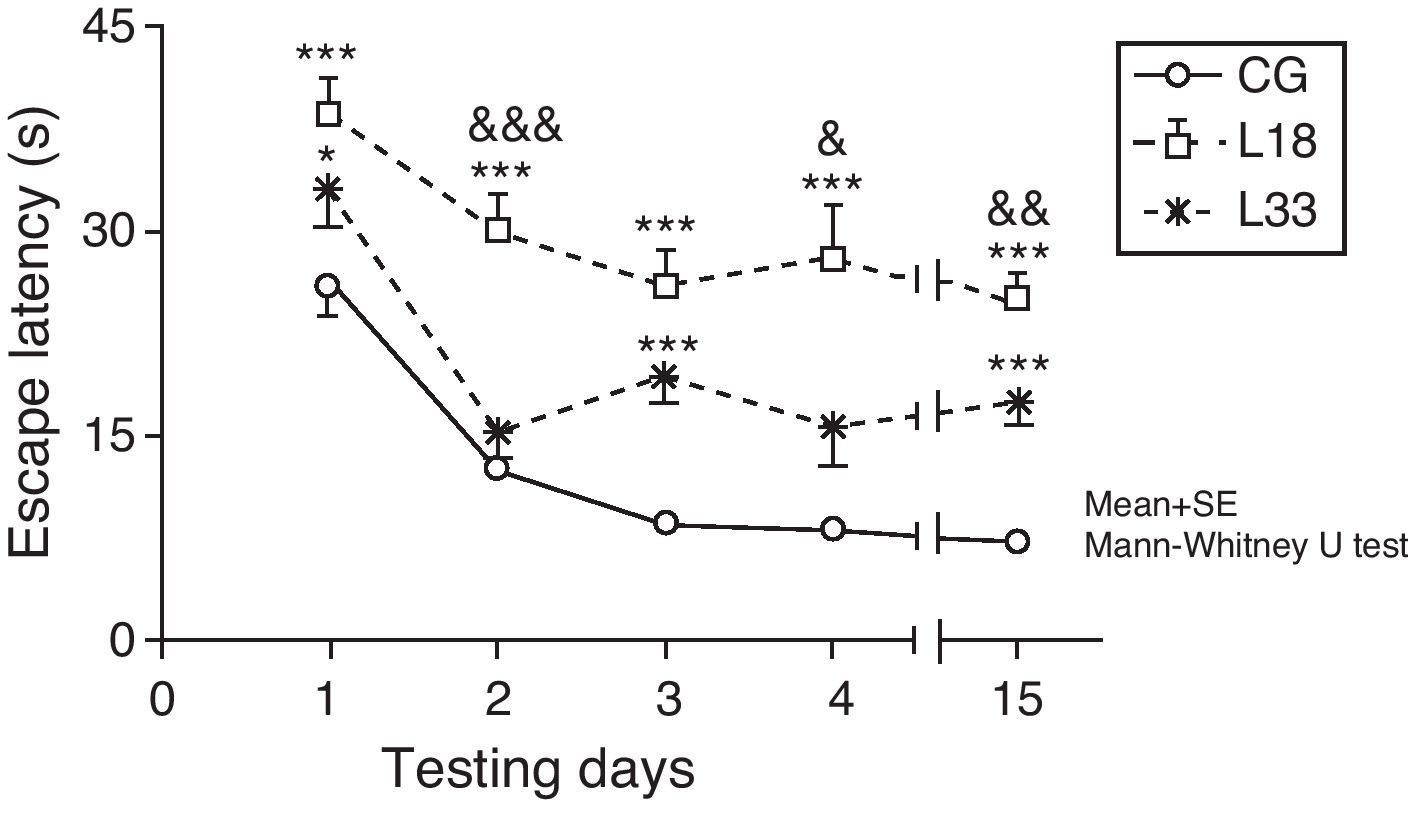
A long-term memory assessment revealed that animals in the study decreased their escape latency period as the days passed (P<.05). Performance by rats with cerebral hypoperfusion at 18 days post-lesion differed significantly from that of control-group animals (P<.001). Escape latency in animals at 33 days post-lesion was longer than in control-group animals and the differences were statistically significant for the first, third, and last testing days (P<.05). Therefore, animals with cerebral hypoperfusion in earlier post-lesion stages showed escape latencies that were significantly longer than animals at 33 days post-lesion on the second, fourth, and last testing days (P<.05) (Fig. 6).

Evaluating long-term or spatial reference memory. In this trial, the platform remained at the same location, but was hidden. Values show mean escape latencies±standard error of the mean. *P<.05: ***P<.001: escape latencies from both model groups compared to control group. &P<.05; &&P<.01; &&&P<.001; differences between escape latencies in both model groups. CG: control group; L18 and L33: animals that began testing at 18 days and 33 days post-lesion.
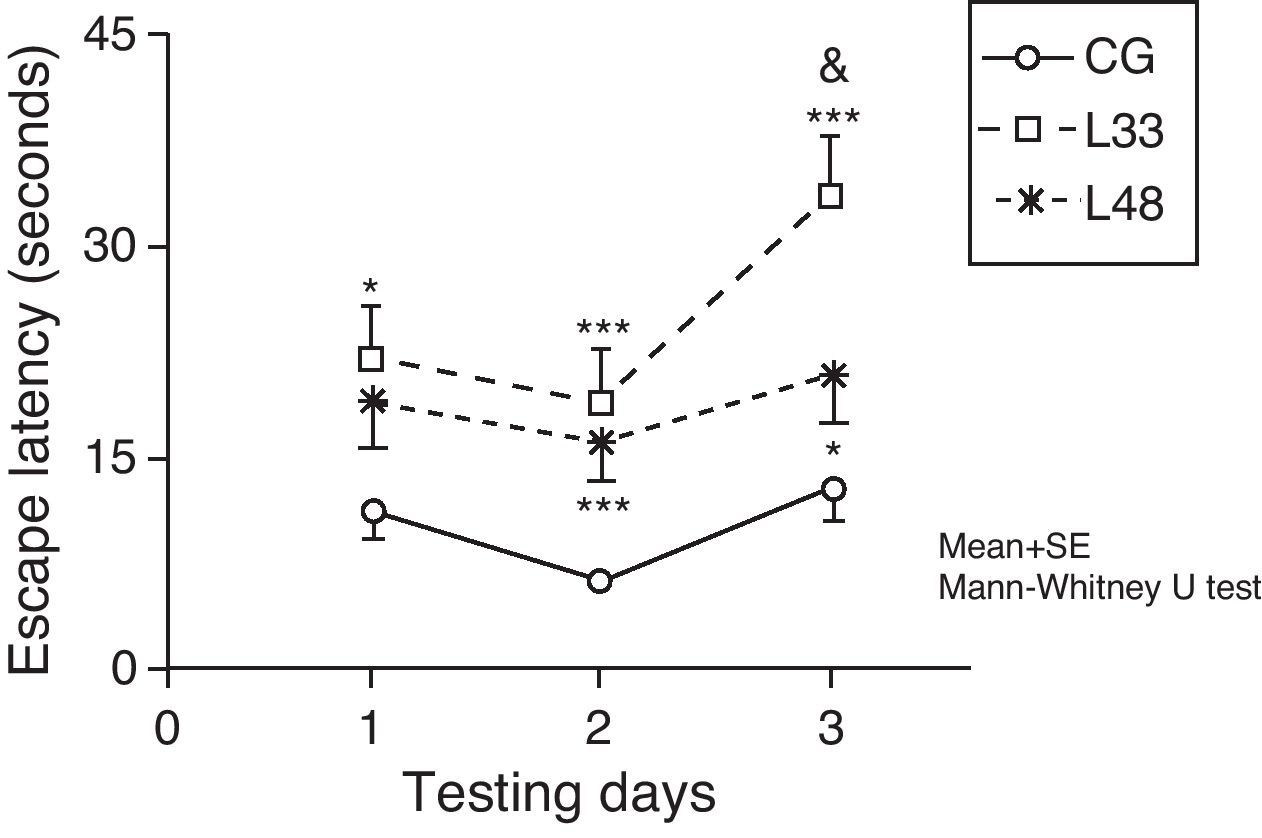
Escape latencies in rats at 33 days post-lesion was significantly longer than latencies in the mock-lesion group for all 3 testing days (Fig. 7) (P<.05). Animals evaluated at 47 days post-lesion showed significant differences in the final 2 testing days (P<.05). Comparison of escape latencies among the model groups revealed that latencies were shorter in the group at the most advanced post-lesion stage. However, this difference was only statistically significant for testing day 3 (P<.05).

Short-term memory impairments as a result of chronic cerebral hypoperfusion. Points along the curve correspond to mean values±standard error of the mean. *P<.05; ***P<.001: indicates that escape latencies in the model group were longer than in the control group. &P<.05: shows differences between different model groups. CG: control group; L33 and L48: animals that began testing at 33 days and 48 days post-lesion.
In ischaemic and oligaemic processes, the generation of reactive oxygen species takes place as a result of decreased blood flow to the brain.12,13 Our studies show that SOD activity did not change in response to acute cerebral hypoperfusion; however, the enzyme was extremely sensitive to chronic cerebral hypoperfusion at 22 days post-lesion. At 24hours post-lesion, the 3 areas of the brain studied here experienced a marked decrease in blood flow,14 which was accompanied by an abrupt decrease in oxygen supply. This fact, added to reperfusion in this model being gradual and very slow, results in the production of only small amounts of superoxide radicals. It is therefore likely that existing SOD is sufficient to eliminate those superoxide radicals. However, at 22 days after POCCA was induced, most of the cerebral blood flow had been restored. As a result, the brain had a better oxygen supply and increased formation of superoxide radicals, which was sufficient to induce SOD activity.
The protective effect of SOD can only be achieved with the consecutive participation of another 2 enzymes that degrade H2O2: CAT and GPX.15 In our study, the increase in CAT activity was only significant in the cortex 24hours after the lesion. This increase in CAT activity took place despite the lack of a parallel increase in SOD activity, which would normally occur with reperfusion.5 This behaviour can be explained if we consider the possible presence of an accumulation of H2O2 which would be accompanied by an increase in CAT activity. If H2O2 is able to accumulate, it may demonstrate that this molecule is stable and able to cross membranes. Furthermore, since reperfusion does not take place suddenly, circulating H2O2 is not eliminated and continues accumulating.
Despite the marked increase in SOD activity in the 3 areas of the brain we studied, we did not identify any significant increases in CAT activity at 22 days post-lesion. Although some in vitro studies show that CAT is essential for intraneuronal H2O2 detoxification,16 the activity of this enzyme in the nervous system has been proven to be very low.17 It is therefore likely that GPX metabolises most of the H2O2 that forms as a product of SOD activity.
The haematoxylin and eosin stain is the most commonly used staining method in histology. The extensive vacuolisation observed in the cortical tissue and striatum of animals that were euthanised 1 week after POCCA resembles that described in other studies published in the scientific literature.18,19 Furthermore, we observed a decrease in neuronal density in the striatum. Neuronal loss in the CA1 and CA3 cell populations of the hippocampus in our study, induced by cerebral hypoperfusion, is coherent with results from other studies.20–22
The reactive astroglioisis found in the cortex of rats with cerebral hypoperfusion supports findings from the study by Schmidt-Kastner et al.23 These authors believe that reduced blood flow in the cortex results in a deficient supply of substrates to the neurons, and that neurons may then induce an astrocyte response. On the other hand, reactive gliosis may occur in answer to a neuronal loss that favours the stimulation of restorative processes in brain tissue, such as the synthesis and release of neurotrophic factors.24 In keeping with this line of reasoning, we discovered a considerable increase in both the size and the number of glial cells in the striatum of the injured rats.
The decrease in GFAP expression associated with the CA1 and CA3 cell populations in the hippocampus corresponds to findings published previously in the literature.25 Astrocytes present different responses to ischaemic events depending on damage magnitude and duration.26 Some have theorised that astrocytes proliferate during early stages of ischaemia, but when the period of hypoperfusion is prolonged, they suffer progressive degeneration.27
The rat model of cerebral hypoperfusion has mainly been associated with neurodegeneration of the hippocampus. The escape latencies for the lesion group in the trial with the visible platform at 22 days post-lesion suggest an impaired ability to locate visual stimuli. This hypothesis is supported by a number of studies showing that cerebral hypoperfusion results in degeneration of the optic tract, retinopathy, and loss of pupillary reflex.7,22,28
Animals with chronic cerebral hypoperfusion at 37 days post-lesion had no difficulty in locating visual stimuli, as demonstrated by the lack of significant differences between the latency periods of animals in the experimental and control groups. This indicates that visual impairment improves as time elapses following the lesion. This finding may be associated with the activation of adaptive mechanisms that restore blood flow levels. Scientists have demonstrated that cerebral hypoperfusion leads to an increase in diameter in the arteries linked to the circle of Willis, which include the basilar artery, the posterior cerebral artery, and the posterior communicating artery.23,29
Analysis of results from the MWM tasks evaluating spatial reference memory reveals that animals in both groups were able to learn the location of the platform. This is corroborated by the significant decrease in escape latency over the days during the testing period. Our results indicate that chronic reduction of cerebral blood flow compromises spatial reference memory, as other authors have also concluded.25,30,31 We must not lose sight of the fact that the animals’ performance on MWM tasks depends on their being able to locate visual clues outside of the maze itself. This is why the visual system impairment occurring in this model elicits poorer performance of this task. However, in this study we did observe an improvement in spatial reference memory among animals in the model group as post-lesion time elapsed. Improvements were parallel to those observed for the visible platform task. This finding suggests that, in tests performed after a longer period of oligaemia, behavioural results may reflect changes in memory and learning processes.
POCCA also induces changes in short-term (working) memory. Animals tested after a longer period of oligaemia performed better than those tested after a shorter period. We feel that this behaviour is due to results from tests in earlier post-lesion phases being affected by the animals’ visual impairment.
In the tests performed by animals that had experienced oligaemia for longer periods, the increases in escape latency were only significant on the second and third testing days. This behaviour is associated with the time elapsed between one trial and the next for each of the testing days. On the first day, the intertrial interval was only 15seconds, meaning that the animal had less difficulty finding the platform.
One hypothesis is that the formation of free radicals is a mechanism contributing to neural impairment.32 Our study demonstrates the existence of an oxidative imbalance in response to chronic cerebral hypoperfusion in the hippocampus, striatum, and cortex. These regions are especially vulnerable to the generation of ROS.33 This finding suggests that the formation of free radicals may be partially responsible for the ongoing cellular damage which characterises the areas of the brain studied in our experimental model.7
On the other hand, we also know that cognitive functions are housed in specific neural circuits which in some cases may span wide areas of the brain. A number of cortical zones, together with the amygdala and the striatum, participate in gathering the information. Some of this information is transferred to the hippocampus. The CA1 and CA3 areas participate in creating short-term memories and transfer this information to the neocortex, which stores it for longer time periods.34 On this basis, we might say that neuropathological changes associated with cerebral hypoperfusion in the POCCA model underlie the deterioration of memory and spatial learning processes.
Animal models for cerebral ischaemia began to be used in the 1970s. The purpose of such models was to enable study of damage caused by cerebral ischaemia under physiologically controlled, repeatable conditions.35
The pathophysiological processes in cerebral ischaemia result from a series of cellular and molecular phenomena taking place over both the short-term and the long-term. They converge in 2 modalities of cell death: necrosis and apoptosis (programmed cell death). Our understanding of these mechanisms is growing, and this is fundamental to the implementation of neuroprotective strategies in clinical practice. Basic knowledge of the pathophysiology of ischaemia and of microglial and macroglial responses is necessary in order to plan neuroprotective strategies. Such strategies must be designed to prevent both acute cell death and later-onset cell death modalities, and also strengthen surviving tissue. Promising neuroprotective drugs are becoming available, and they are being studied in both experimental animal models and in clinical trials in humans.2
Different phases have been suggested for neuroprotective strategies, depending on which molecular event is the target of the intervention. Experimental studies enabling molecular characterisation and establishing time windows for such interventions are available and may be consulted for further information. Neuroprotective strategies are based on detailed knowledge of each of these phases, and scientists search for key events that may be targeted by pharmacological or physical interventions aimed at limiting neuronal damage and facilitating recovery.2 With this in mind, the rat model of induced cerebral hypoperfusion may be useful for evaluating the efficacy of neurorestorative, neuroprotective, and antioxidant therapeutic strategies that promote recovery of cognitive functions.
The main purpose of studies carried out using experimental models of cerebral ischaemia must be to compile basic knowledge about pathobiological processes underlying ischaemic injury. These studies should not be used merely to demonstrate treatment benefits as a preliminary step in the design of clinical trials.35
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Castaño Guerrero Y, et al. Alteraciones del metabolismo oxidativo y de la memoria y el aprendizaje en un modelo de hipoperfusión cerebral en ratas. Neurología. 2013;28:1–8.
