



Hereditary neuropathy with liability to pressure palsy (HNPP) is an autosomal dominant disorder, typically presenting with recurrent episodes of mononeuropathy in nerves susceptible to compression, with similar neurophysiological characteristics. However, other clinical and neurophysiological presentations have been reported.
MethodsWe retrospectively analysed the clinical and neurophysiological characteristics of 20 patients with genetically confirmed HNPP. Sixteen patients were studied in our department between 1996 and 2016.
ResultsIn addition to the typical characteristics of HNPP, we found atypical forms including recurrent positional sensory symptoms in 3 patients, chronic sensorimotor polyneuropathy in one, and non-progressive mononeuropathy in one. Onset was early in 2 patients: one at the age of 7 years, with common peroneal nerve injury, and another at birth, with brachial plexus involvement. By frequency, the main pathological findings in the nerve conduction study were: decreased sensory nerve conduction velocity in the sural (84%) and the median and superficial peroneal nerves (94%); decreased motor nerve conduction velocity in the ulnar nerve through the elbow (97%), and increased motor distal latency of the median and deep peroneal nerves (74%).
ConclusionOur results confirm the clinical variability of HNPP, with the most frequent nerve conduction study findings being the generalised decrease in sensory nerve conduction velocity, in addition to motor involvement, mainly in locations susceptible to nerve compression. The nerve conduction study can detect typical, atypical, and asymptomatic cases of HNPP.
La neuropatía hereditaria con parálisis sensible a la presión (NHPP) es una alteración autosómica dominante, con episodios recurrentes de mononeuropatía en nervios susceptibles de compresión típicos, con características neurofisiológicas comunes. Sin embargo, se han comunicado otras presentaciones clínicas y neurofisiológicas.
MétodosAnálisis de características clínicas y neurofisiológicas en una revisión retrospectiva de 20 pacientes con NHPP confirmados genéticamente, de los cuales, 16 se estudiaron en nuestro servicio entre los años 1996 y 2016.
ResultadosAdemás de las características típicas de la NHPP, encontramos formas atípicas como síntomas sensitivos recurrentes posicionales en 3 pacientes, polineuropatía sensitivo motora crónica en uno y mononeuropatía no evolutiva en otro. Dos pacientes empezaron en edad temprana, uno a los 7 años, con lesión del ciático poplíteo externo y otro al nacer, con afectación de plexo braquial. Las principales alteraciones de la conducción nerviosa que se encontraron fueron: la disminución de la velocidad de conducción sensitiva con rangos del 84% en el sural y 94% en el mediano y peroneo superficial, descenso de la velocidad de conducción motora del nervio cubital a través del codo en el 97% y el incremento de la latencia distal motora del nervio mediano y ciático poplíteo externo en el 74%.
ConclusiónDe acuerdo con los resultados, confirmamos la variabilidad clínica de la NHPP y encontramos los hallazgos más frecuentes en la conducción nerviosa, la disminución generalizada de la velocidad de conducción sensitiva, además de la afectación motora principalmente en lugares susceptibles de compresión, y detectamos los casos típicos, atípicos y asintomáticos de esta en entidad.
Hereditary neuropathy with liability to pressure palsies (HNPP) is a peripheral neuropathy with onset in the second or third decades of life. Few cases of early-onset HNPP have been reported. The condition typically presents with acute, recurrent episodes of non-painful nerve palsy at common sites of nerve entrapment following minor trauma or compression. Most patients recover completely. Atypical clinical presentations have been reported, including non-progressive mononeuropathy,1,2 chronic sensorimotor polyneuropathy, progressive sensorimotor mononeuropathy,3 multifocal compression neuropathy, Charcot-Marie-Tooth disease-like chronic progressive polyneuropathy,4 bilateral acute radial nerve palsy, short-term recurrent sensory symptoms, and scapuloperoneal syndrome.5
The exact pathophysiological mechanism is unknown. Most cases are associated with a 1.5Mb deletion in the 17p11.2 region containing the peripheral myelin protein 22 (PMP22) gene.6–8 In some cases, mutations in PMP22 may be found.4
The disease is underdiagnosed due to its benign course2,3 and the fact that some patients are asymptomatic.9–12 Neurophysiological studies are able to detect not only patients with typical or atypical manifestations,13–21 but also asymptomatic relatives, which helps in identifying carriers of the gene.2,19 Better understanding of the neurophysiological characteristics of HNPP is extremely valuable. The most frequent findings are motor conduction alterations,4,12,16,22,23 mainly at common sites of compression, including increased distal motor latency (DML) of the median nerve, decreased motor conduction velocity (MCV) of the ulnar nerve at the elbow, and increased DML or decreased MCV in at least one of the external popliteal sciatic nerves (EPSN). Less frequently, cases have been reported of sensory nerve conduction alterations,22,24,25 such as decreased sensory amplitude in the upper limbs or a generalised decrease in sensory conduction velocity (SCV). Furthermore, some authors have described a pattern of sustained alterations consisting of decreased SCV in the wrist-palm segment of both median nerves.3,13
In view of the wide range of presentations of HNPP, we analysed the neurophysiological and clinical characteristics of all patients with 17p11.2 deletions attended at our centre over a 20-year period.
Material and methodsWe conducted a retrospective analysis of the medical histories of 20 patients presenting deletions in chromosome 17p11.2 affecting PMP22 and attended at Complejo Hospitalario de Navarra between 1996 and 2016. The study complies with the ethical standards of our centre's ethics committee.
We gathered the following data from medical histories: family history of the disease vs isolated case, age at symptom onset, clinical manifestations, clinically affected nerves, physical examination results, and progression.
Neurophysiological studies were performed in 16 patients using a Synergy 5-channel EMG system and software (version 15.0; Natus Inc.), according to standard protocols.26,27 Skin temperature was kept above 31°C during the study. Stimulation and signal recording were performed with surface electrodes.
Motor nerve conduction was studied in the median, ulnar, EPSN, and posterior tibial nerves, and recorded in the abductor pollicis brevis, abductor digiti minimi, extensor digitorum brevis, and abductor hallicus muscles, respectively. DML was obtained with stimulation at a distance of 3cm for the medial and ulnar nerves, 5cm for the EPSN, and 8cm for the posterior tibial nerve. MCV was measured in the following segments: wrist to elbow for the median nerve, wrist to above the elbow for the ulnar nerve, and ankle to popliteal fossa for the posterior tibial nerve and EPSN. We studied additional segments at common sites of compression: the elbow for the ulnar nerve and the head of the fibula for the EPSN. A decrease of 10m/s or more in MCV was considered a conduction alteration in these segments; a 30% reduction in compound muscle action potential amplitude (proximal vs distal values) was regarded as motor nerve conduction block.
We used orthodromic techniques to study sensory nerve conduction in the upper limbs, in the median (from the proximal phalanx of the second digit to the wrist) and ulnar nerves (from the proximal phalanx of the fifth digit to the wrist). In the lower limbs, antidromic techniques were applied to assess sensory nerve conduction of the sural nerve (from mid-calf to lateral malleolus) and the superficial peroneal nerve (from the lateral distal third of the leg to the midpoint between the lateral malleolus and the anterior tibial tendon). Sensory nerves were stimulated at a distance of 14cm in the sural nerve and 10cm in the superficial peroneal nerve; SCV was measured at peak amplitude.
We used the reference values of our neurophysiology laboratory and other centres.26–28
ResultsPatients and family historyData were gathered from 20 patients with 7p11.2 deletion: 11 women and 9 men. Age at symptom onset ranged from 0 to 74 years. Eighteen patients (90%) belonged to 7 families with history of HNPP, which followed an autosomal dominant inheritance pattern. Only 2 patients (10%) were apparently sporadic cases, with no known family history of the condition.
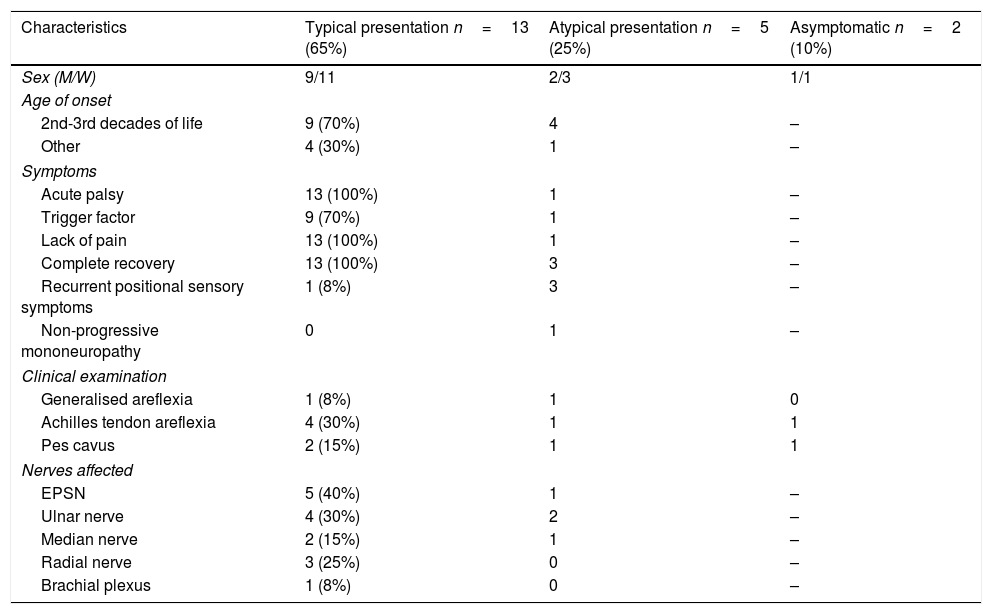
Presentation and clinical characteristicsTwo patients (10%) were asymptomatic, 13 (65%) presented typical clinical characteristics, and the remaining 5 (25%) presented atypical symptoms (Table 1).
Clinical characteristics of our sample, by form of presentation (typical, atypical, or asymptomatic).
| Characteristics | Typical presentation n=13 (65%) | Atypical presentation n=5 (25%) | Asymptomatic n=2 (10%) |
|---|---|---|---|
| Sex (M/W) | 9/11 | 2/3 | 1/1 |
| Age of onset | |||
| 2nd-3rd decades of life | 9 (70%) | 4 | – |
| Other | 4 (30%) | 1 | – |
| Symptoms | |||
| Acute palsy | 13 (100%) | 1 | – |
| Trigger factor | 9 (70%) | 1 | – |
| Lack of pain | 13 (100%) | 1 | – |
| Complete recovery | 13 (100%) | 3 | – |
| Recurrent positional sensory symptoms | 1 (8%) | 3 | – |
| Non-progressive mononeuropathy | 0 | 1 | – |
| Clinical examination | |||
| Generalised areflexia | 1 (8%) | 1 | 0 |
| Achilles tendon areflexia | 4 (30%) | 1 | 1 |
| Pes cavus | 2 (15%) | 1 | 1 |
| Nerves affected | |||
| EPSN | 5 (40%) | 1 | – |
| Ulnar nerve | 4 (30%) | 2 | – |
| Median nerve | 2 (15%) | 1 | – |
| Radial nerve | 3 (25%) | 0 | – |
| Brachial plexus | 1 (8%) | 0 | – |
The 13 patients with typical manifestations presented recurrent, regressive acute palsy triggered by a precipitating factor, and recovered within days or months; the affected nerves were the EPSN in 40% of cases, the ulnar nerve in 30%, the radial nerve in 25%, the median nerve in 15%, and the brachial plexus in 8%. Symptom onset occurred between the second and third decades of life in 9 patients, after the third decade of life in 2, and at very young ages in the remaining 2. One of these 2 patients presented symptoms after birth due to trauma caused by a forceps (brachial plexopathy due to proximal weakness in the right upper limb), but recovered completely after several months of rehabilitation therapy. This patient also presented generalised hypotonia and intellectual disability. The patient's father, 2 paternal uncles, and a cousin also presented HNPP. Diagnosis was confirmed by genetic analysis and a neurophysiological study conducted at the age of one year. At the time of writing (age 15 years), the patient has presented no further episodes of palsy. The second patient with symptom onset during childhood presented clinical manifestations at the age of 7 years, with pes cavus and weakness affecting the right EPSN, and recovered within days. His mother, 3 maternal uncles, and his maternal grandmother also had HNPP. To date (age 23), he has presented no further episodes of palsy; he underwent surgery for pes cavus at 20 years of age.
All 5 patients with atypical clinical manifestations had family history of HNPP; age at onset ranged from 24 to 60 years, with 80% presenting the condition between the second and third decades of life. Three patients presented recurrent, short-term positional sensory symptoms in the territories of the median and ulnar nerves. In another patient, HNPP manifested as chronic sensorimotor polyneuropathy. The patient consulted at the age of 60 years due to chronic generalised paraesthesia and weakness; the examination revealed generalised areflexia, pes cavus, and peroneal muscle atrophy. She had 2 children diagnosed with HNPP. The last patient with atypical symptoms presented the initial manifestations at the age of 24. The disease started with classic symptoms: upper and lower limb palsy, involving the EPSN and the ulnar nerve, during sleep and in certain positions. Symptoms resolved within days and reappeared every 1-2 years. At the age of 31, he presented an atypical episode with left EPSN palsy, which did not resolve and progressed to non-progressive chronic mononeuropathy. At present, weakness in foot dorsiflexion persists. Although nerve conduction studies initially presented findings compatible with HNPP, genetic studies yielded no conclusive data; at the age of 65 years, diagnosis was confirmed by genetic analysis.
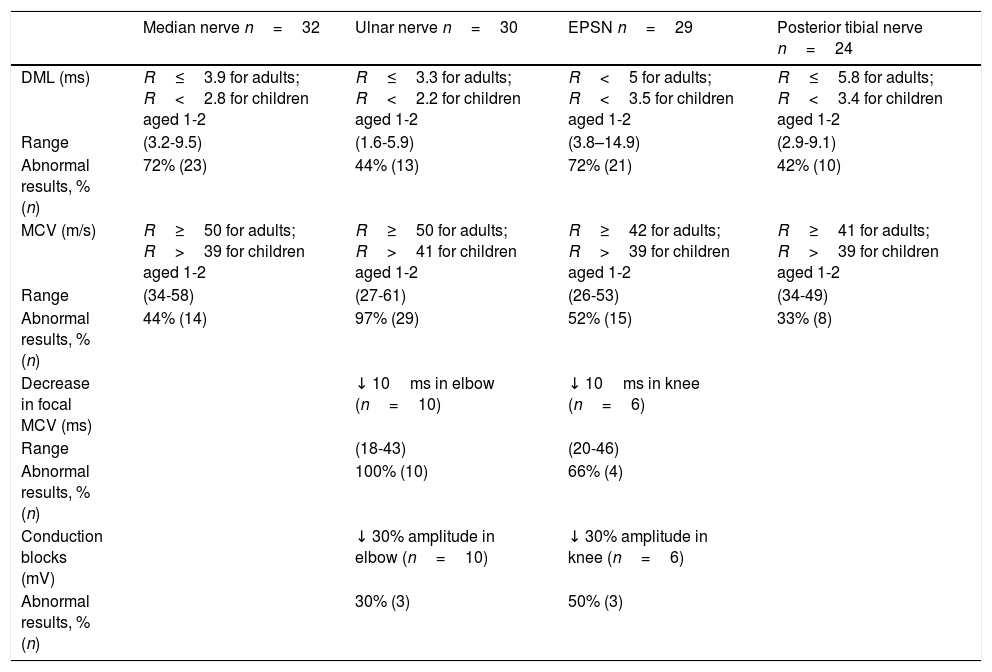
Neurophysiological studyNeurophysiological studies were performed in 16 patients: 12 with typical symptoms, 2 with atypical symptoms, and the 2 asymptomatic patients (Tables 2 and 3). The ages of our patients at the time of the neurophysiological study ranged from 14 months to 76 years.
Neurophysiological study of 16 patients with 1.5Mb deletion in chromosome 17p11.2: motor conduction parameters.
| Median nerve n=32 | Ulnar nerve n=30 | EPSN n=29 | Posterior tibial nerve n=24 | |
|---|---|---|---|---|
| DML (ms) | R≤3.9 for adults; R<2.8 for children aged 1-2 | R≤3.3 for adults; R<2.2 for children aged 1-2 | R<5 for adults; R<3.5 for children aged 1-2 | R≤5.8 for adults; R<3.4 for children aged 1-2 |
| Range | (3.2-9.5) | (1.6-5.9) | (3.8–14.9) | (2.9-9.1) |
| Abnormal results, % (n) | 72% (23) | 44% (13) | 72% (21) | 42% (10) |
| MCV (m/s) | R≥50 for adults; R>39 for children aged 1-2 | R≥50 for adults; R>41 for children aged 1-2 | R≥42 for adults; R>39 for children aged 1-2 | R≥41 for adults; R>39 for children aged 1-2 |
| Range | (34-58) | (27-61) | (26-53) | (34-49) |
| Abnormal results, % (n) | 44% (14) | 97% (29) | 52% (15) | 33% (8) |
| Decrease in focal MCV (ms) | ↓ 10ms in elbow (n=10) | ↓ 10ms in knee (n=6) | ||
| Range | (18-43) | (20-46) | ||
| Abnormal results, % (n) | 100% (10) | 66% (4) | ||
| Conduction blocks (mV) | ↓ 30% amplitude in elbow (n=10) | ↓ 30% amplitude in knee (n=6) | ||
| Abnormal results, % (n) | 30% (3) | 50% (3) |
DML: distal motor latency; MCV: motor conduction velocity; n: number of nerves registered; R: reference value.
Neurophysiological study of 16 patients with 1.5Mb deletion in chromosome 17p11.2: sensory conduction parameters.
| Median nerve n=32 | Ulnar nerve n=31 | Sural nerve n=26 | Superficial peroneal nerve n=17 | |
|---|---|---|---|---|
| SCV (m/s) | R>50 for adults; R>46 for children aged 1-2 | R>50 for adults; R>38 for children aged 1-2 | R≥41 for adults; R>40 for children aged 1-2 | R>40 for adults; R>39 for children aged 1-2 |
| Range | (28-52) | (29-54) | (25-41) | (21-41) |
| Abnormal results, % (n) | 94% (30) 3 NR | 90% (28) 3 NR | 84% (22) 3 NR | 94% (16) 3 NR |
| Sensory amplitude (μV) | R>8 for adults; R>7 for children aged 1-2 | R≥7 for adults; R>13 for children aged 1-2 | R>6 for adults; R>8 for children aged 1-2 | R≥6 for adults; R>8 for children aged 1-2 |
| Range | (0.4-11) | (0.6-7) | (1-11) | (2-9) |
| Abnormal results, % (n) | 78% (25) 3 NR | 70% (22) 3 NR | 38% (10) 4 NR | 41% (7) 3 NR |
n: number of nerves registered; NR: nerves with no response; R: reference values; SCV: sensory conduction velocity.
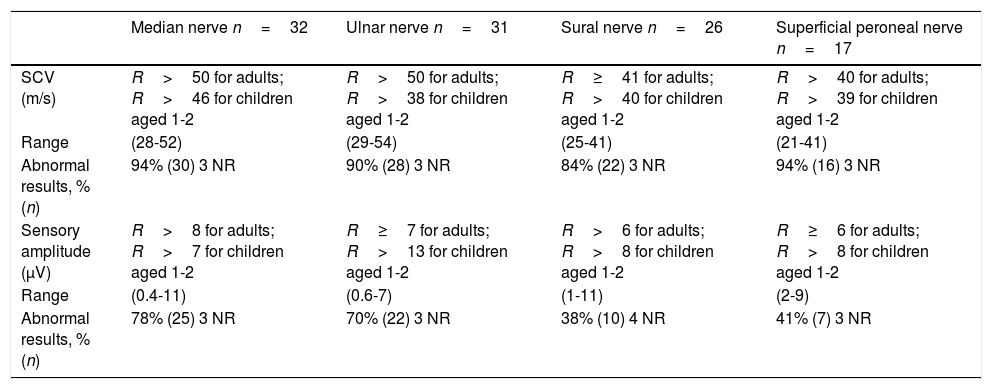
Sensory nerve conduction was markedly impaired in our sample, with high rates of SCV alteration in all the nerves studied, ranging from 84% (sural nerve) to 94% (median and superficial peroneal nerves). Decreased sensory amplitude was detected in the median nerve in 28% of patients, in the ulnar nerve in 70%, in the superficial peroneal nerve in 41%, and in the sural nerve in 38%.
Among motor and sensory conduction parameters, the most frequent alteration was decreased MCV of the ulnar nerve at the elbow (97% of patients), followed by increased DML of the median nerve and the EPSN (72% in both cases). Other findings included decreased MCV in the EPSN (52%), the median nerve (44%), and the posterior tibial nerve (33%), and increased DML in the ulnar (44%) and posterior tibial nerves (42%).
Short-segment nerve conduction studies of the ulnar nerve (at the elbow) and the EPSN (at the head of the fibula) were only performed in 6 and in 3 patients, respectively. Conduction in these segments was decreased in 100% of patients in the ulnar nerve and 66% in the EPSN; 30% of ulnar nerves and 50% of EPSNs presented conduction blocks.
DiscussionIn our series, 2 patients presented HNPP at unusual ages. Both had family history of the condition and presented the initial manifestations at very young ages: at 7 years, in the form of EPSN palsy, and at birth, with hypotonia and brachial plexopathy due to trauma during birth. Both progressed favourably, having presented no further episodes of palsy at the time of writing, and were diagnosed at an early age based on results from neurophysiological studies and genetic analyses. Our literature search only found 3 reported cases of HNPP with onset at birth. One patient presented symptoms of transient Erb palsy,8 as in one of our patients; another presented peroneal atrophy and pes cavus9; and the third patient presented EPSN palsy at birth and recovered completely (diagnosis was established at age 7 years due to muscle weakness and hypotonia).10
From a clinical viewpoint, the most frequently affected territories were the EPSN and the ulnar nerve, and to a lesser extent the median and radial nerves, with only one case of brachial plexopathy. This stands in contrast with other published studies reporting that the EPSN, ulnar nerve, and brachial plexus are the most frequently affected territories.4,12,13 Brachial plexopathy may be recurrent and is typically associated with other symptoms. Presentation of brachial plexopathy as the sole manifestation of HNPP, as in our patient, is rare.14 Isolated cases have been reported featuring involvement of the facial, trigeminal, hypoglossal, and recurrent laryngeal nerves.15 None of our patients presented involvement of any of these nerves, however.
Regarding the form of presentation, 5 of our patients (25%) showed atypical manifestations. This frequency is similar to those described in other series.4,13 The different atypical forms of HNPP were classified by Pareyson et al.,16 in 1996, and by Mouton et al.,13 in 1999, into 5 groups: (1) recurrent positional sensory symptoms; (2) progressive form with EPSN alterations; (3) chronic sensorimotor polyneuropathy; (4) Charcot-Marie-Tooth disease-like sensorimotor polyneuropathy; and (5) chronic demyelinating polyradiculoneuropathy. In 2002, Pou Serradell et al.4 added chronic truncal compression syndrome (involving multiple channels), non-progressive chronic monotruncal weakness syndrome, and monomelic distal atrophy. Cases have also been described of central nervous system involvement, with reduced white matter volume and cognitive impairment,15 hereditary sensorimotor neuropathy type VIII (polyneuropathy associated with cerebellar-extrapyramidal syndrome),17 and the case of a 7-year-old girl with gait alterations, pes cavus, scoliosis, and torticollis, who presented no episodes of palsy.18 Atypical presentations in our series comprised recurrent sensory symptoms in 3 patients, chronic sensorimotor polyneuropathy in one, and non-progressive mononeuropathy in another patient. Interestingly, we observed different forms of disease presentation in individuals from the same family, with one patient presenting recurrent sensory symptoms, another displaying non-progressive mononeuropathy, and the third presenting typical manifestations of HNPP. This great phenotypic variability between family members is unusual.19–21
Regardless of the clinical presentation, and including the 2 asymptomatic patients, all the patients who underwent neurophysiological studies showed nerve conduction alterations. In our patients, sensory involvement was more severe than motor involvement (84%-94% vs 33%-97%). Other series report more predominant motor involvement4,12,16,22,23,29 or, less frequently, predominant sensory involvement.22,24,25 This variability may be due to the small size of the samples24,25 or the small number of sensory nerves evaluated.2,8,13 In addition to the commonly evaluated sensory nerves (median, ulnar, and sural nerves), our neurophysiological protocol also explores the superficial peroneal nerve; this may explain the higher percentage of sensory involvement in our series.
Motor conduction alterations were mainly observed in common sites of compression. The most frequently affected motor conduction parameters were MCV of the ulnar nerve at the elbow and DML of the median nerve and the EPSN. These findings are consistent with the results of most published studies.2,3,8,11–13,16,23 A disproportionate increase in DML as compared to MCV has been reported as a typical finding in nerve conduction studies of patients with HNPP.25 This theory is controversial30 as the findings cannot be extrapolated to all motor nerves. Some nerves, such as the median nerve or the EPSN, are likely to display distal slowing due to repetitive trauma.30 In our series, these findings were observed in the median nerve and the EPSN but not in the ulnar or posterior tibial nerves.
Another motor conduction alteration frequently evaluated for diagnosis of HNPP is the presence of conduction blocks at common entrapment sites. Our sample presents higher prevalence of this sign than normal.13 One of our patients presented a subclinical conduction block of the left EPSN, with persistent symptoms, similarly to other cases described in the literature.31 The mechanism responsible for conduction block in HNPP is poorly understood. Axonal excitability studies have shown alterations in the electrical stimulation threshold, which are more marked in the wrist and also detectable in the elbow. According to the authors, structural abnormalities at the node of Ranvier may predispose the nerves to conduction block in response to pressure or stretching.32
DNA analysis is a useful objective tool in the diagnosis of HNPP. However, given the hereditary nature of this neuropathy, we recommend performing neurophysiological studies since they not only provide useful additional data from patients with confirmed diagnosis but are also able to detect asymptomatic individuals; the relatives of patients with HNPP should be screened for the condition.2,11
ConclusionGeneralised SCV alterations, decreased MCV of the ulnar nerve at the elbow, and increased DML of the median nerve and the EPSN are the main neurophysiological characteristics of HNPP with predominantly sensory involvement. Understanding the neurophysiology of the disease helps detect patients with the typical clinical symptoms of HNPP, as well as patients with atypical presentations and even asymptomatic individuals. In view of the considerable phenotypic variability of the condition, neurophysiological studies constitute an extremely useful diagnostic tool.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Pabón Meneses RM, Azcona Ganuza G, Urriza Mena J, Ibiricu Yanguas A, Gila Useros L, García de Gurtubay I. Hallazgos clínico-neurofisiológicos en neuropatías hereditarias sensibles a la presión con deleción del cromosoma 17p11.2. Neurología. 2022;37:243–249.
