



Myasthenia gravis (MG) and neuromyelitis optica (NMO) are immune-mediated disorders with low prevalence in the general population. NMO is an inflammatory disease of the central nervous system that mainly affects the spinal cord and optic nerve, and is associated with presence of anti–aquaporin-4 antibodies (AQP4-Ab) in 60%-80% of patients.1 AQP4-Ab–negative patients with compatible symptoms are considered to have neuromyelitis optica spectrum disorders (NMOSD), which also include a subgroup of patients positive for anti–myelin oligodendrocyte glycoprotein antibodies.2 MG is a neuromuscular disorder associated with the presence of anti–acetylcholine receptor antibodies (AChR-Ab) and characterised by fluctuating muscle weakness.3 Although NMO and MG affect the nervous system in different ways, an association between the 2 has been reported in the literature,4 which suggests that they may share some pathogenic mechanism. We present 2 cases of MG associated with NMO.
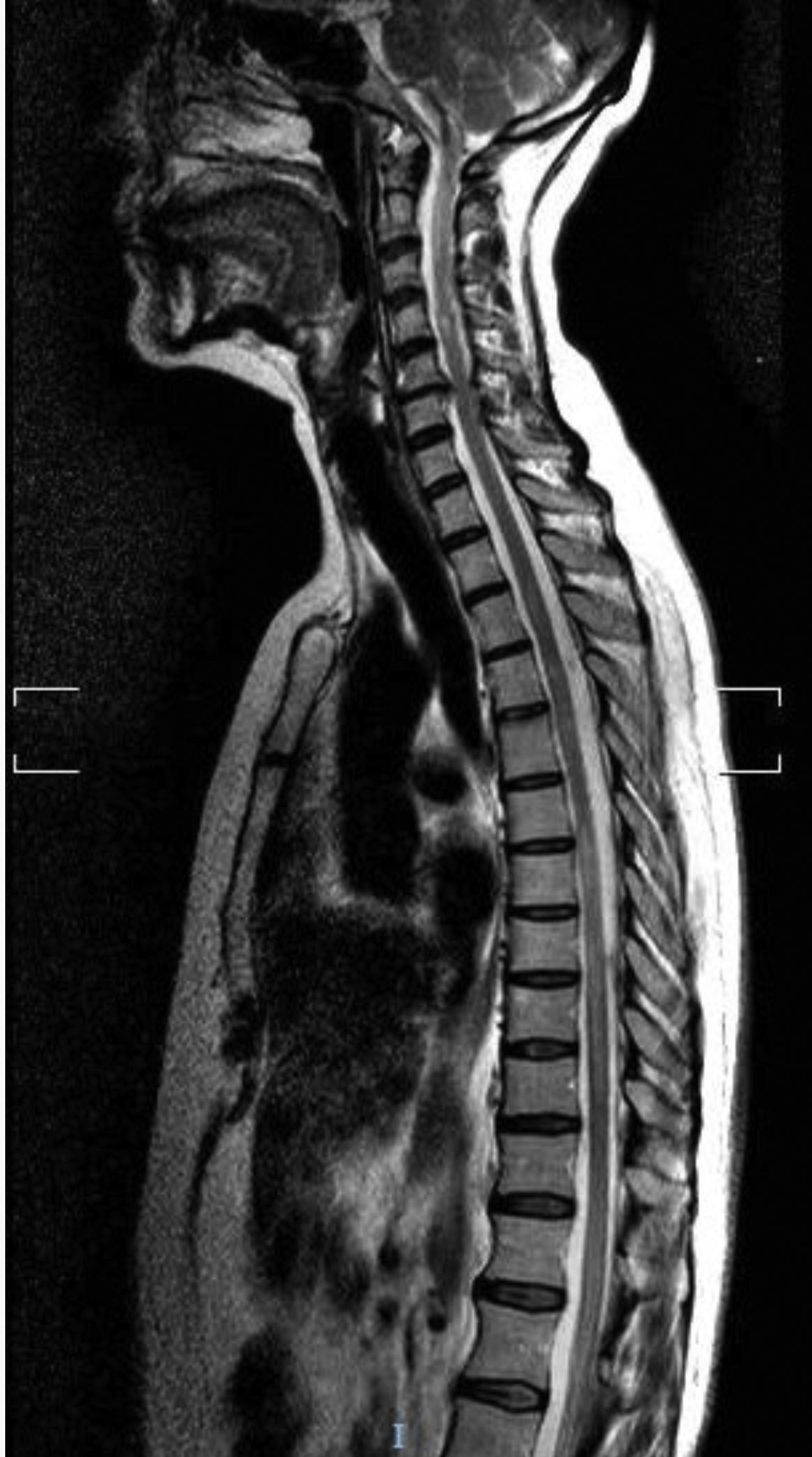
Our first patient was a 49-year-old white woman with AChR-Ab–positive MG not associated with thymoma, diagnosed 6 years previously; she had not undergone thymectomy due to good control of myasthenic symptoms with pyridostigmine and low-dose prednisone. She was admitted to the neurology department due to subacute weakness, right leg hypoaesthesia, and band-like thoracic pain. The neurological examination revealed paresis of the right leg and right-sided hypoaesthesia at the T7 sensory level, lower limb hyperreflexia, and extensor plantar reflex in the right foot. A brain MRI scan revealed a non-specific lesion in the frontal region, and a spinal cord MRI scan revealed minimally expansive hyperintensities at C1-C3, T4-T9, and T11-L2 on T2-weighted sequences (Fig. 1). CSF analysis revealed no abnormalities. The patient tested negative for oligoclonal bands and presented an AQP4-Ab titre of 6.99 (normal range, < 1). The patient was treated with boluses of methylprednisolone, followed by prednisone in decreasing doses; symptoms improved, with the only sequelae being hypoaesthesia in the T7 band and difficulty urinating. She continued receiving low-dose prednisone and mycophenolate mofetil.

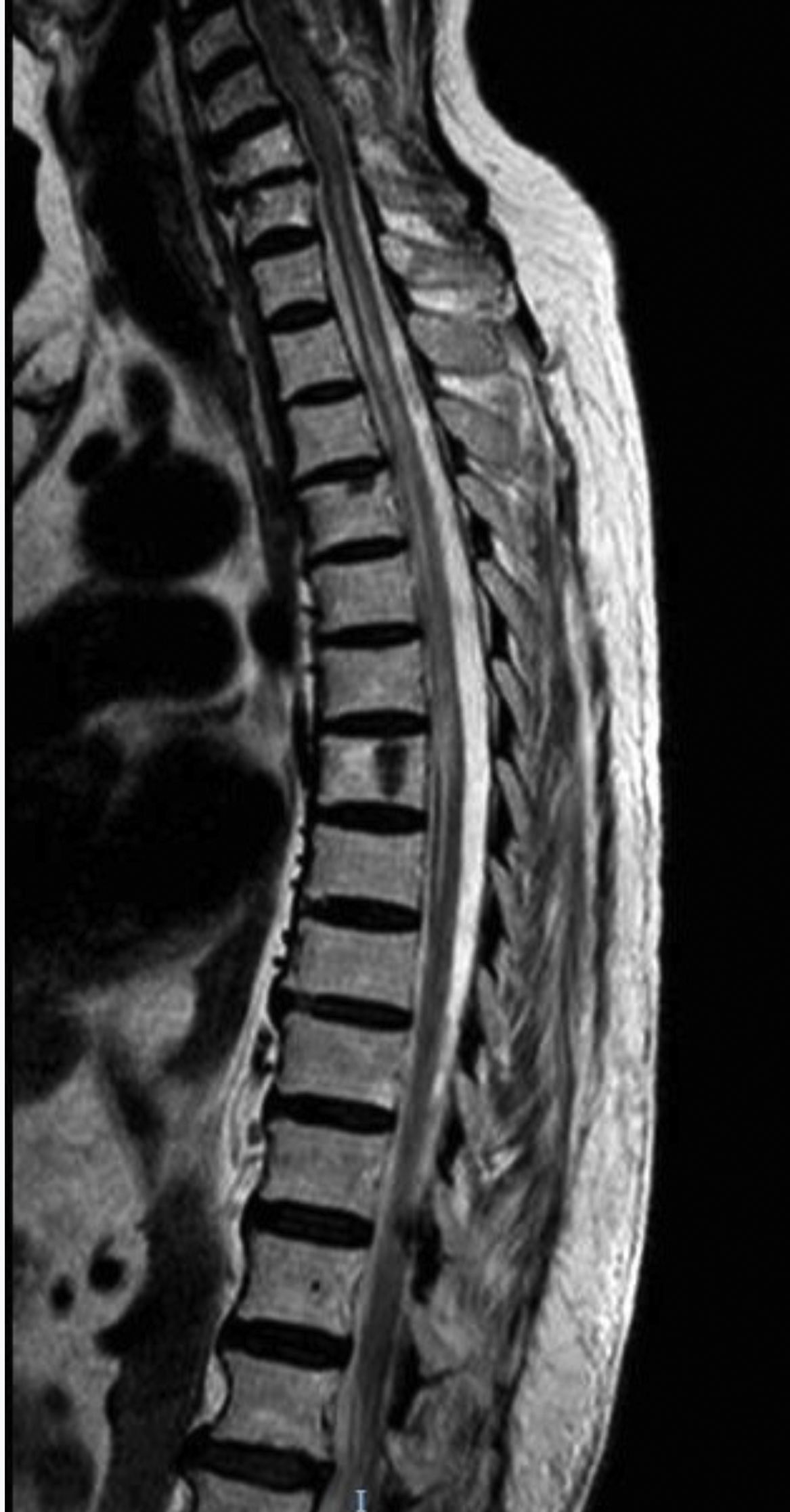
Our second patient was a 76-year-old white woman with type 2 diabetes mellitus, autoimmune hypothyroidism, and Sjögren syndrome. She had history of AChR-Ab–positive ocular MG; symptoms were adequately controlled with pyridostigmine. The patient visited the emergency department due to abdominal pain and ascending lower limb weakness and paraesthesia, and subsequently presented urinary incontinence. The neurological examination revealed paraparesis with overactive reflexes and suspended sensory loss at T3-L4. A T2-weighted MRI scan revealed a hyperintense lesion at C7-T8, with no gadolinium uptake (Fig. 2). A CSF analysis yielded normal results, with negative oligoclonal bands. Laboratory analysis revealed presence of AQP4-Ab in the serum and CSF at a titre of 1:10. An autoimmune study detected antinuclear antibodies and extractable nuclear antigens in the blood. The patient was treated with boluses of methylprednisolone, followed by decreasing doses of prednisone; her symptoms improved, but mild paraparesis persisted. She continued treatment with mycophenolate mofetil and prednisone. Ten months after the initial episode, she presented another episode of myelitis, which was treated with boluses of methylprednisolone, followed by prednisone. Mycophenolate mofetil was suspended and the patient was started on intravenous rituximab dosed at 1 g, with the first 2 doses separated by 2 weeks and subsequent doses separated by 6 months; she has shown no symptom recurrence to date.

NMO is an inflammatory disease frequently associated with presence of AQP4-Ab,1 a highly specific marker of the disease.5 The pathogenicity of these antibodies is thought to be explained by their binding to astrocytes, with subsequent complement activation. Furthermore, patients with MG show IgG1 and IgG3 AChR antibodies, which also activate the complement system, leading to motor endplate dysfunction.6
Copresence of MG and NMO was first reported in 1995; several other cases have since been described. Most patients are non-white women, in whom MG was diagnosed first. In 72.9% of patients, NMO presented after thymectomy, unlike in the 2 cases presented here. Positivity for AChR-Ab and AQP4-Ab was detected in 97.7% and 90% of patients, respectively,3 and several studies indicate that MG was mild, with some patients even presenting remission of MG.4,7 Few data are available on the most appropriate treatment for these patients. Intravenous corticoids and/or plasmapheresis may be used in the acute phase,1,4,7 followed by long-term treatment with different immunosuppressants (corticosteroids, azathioprine, cyclophosphamide, rituximab, mitoxantrone); progression varies between patients, and outcomes are often poor.1,4,7
Both NMO and MG present low prevalence in the general population.8,9 Both conditions may be considered autoimmune channelopathies, and present several similarities: both involve genetic and environmental factors, are more frequent in women, and present in association with other systemic or organ-specific autoimmune diseases.10 Sjögren syndrome and systemic lupus erythematosus, among other autoimmune diseases, have been widely described in patients with MG and NMO. NMO is probably explained by the co-presence of different entities; positivity for anti-AQP4-Ab is highly specific of patients with NMO and symptoms of optic neuritis or longitudinally extensive transverse myelitis.11 In fact, several studies have shown that up to 20%-30% of patients with NMO also present other autoimmune diseases,12 and a case-control study of patients with NMO showed that 2% had MG and 11% presented AChR-Ab.13
Given the low prevalence of both entities, we may expect to observe a lower proportion of patients presenting MG and NMO. However, the association between the 2 entities is unsurprising considering that AQP4 is expressed not only in the central nervous system but also in skeletal muscles14 and thymic epithelial cells.15 The thymic abnormalities occurring in patients with MG may constitute a risk factor for AQP4-Ab production due to AChR-Ab − induced degeneration of the postsynaptic membrane.16 The pathogenesis of this association may be linked to CD4+ regulatory T cells, which are involved in immunologic self-tolerance and whose levels may decrease or be altered in both entities.4,7 The reason why NMO more frequently co-presents in patients with MG undergoing thymectomy is also unknown, although it seems to be linked to the role of the thymus in self-tolerance in adulthood.
In conclusion, we should be aware of the possible association between MG and NMO; correct diagnosis may have considerable implications for treatment and prognosis.
Please cite this article as: Piñar Morales R, Todorova Petrova M, Barrero Hernández FJ. Coexistencia de miastenia gravis y neuromielitis óptica: descripción de dos casos. Neurología. 2021;36:174–176.
