




Longitudinally extensive transverse myelitis (LETM) has classically been grouped with the full or limited neuromyelitis optica spectrum disorders (NMOSD). However, differential diagnosis reveals a wide range of aetiologies.
ObjectiveTo report on differential diagnosis and prognosis for LETM observed in a group of patients in Buenos Aires, Argentina.
Patients and methodsCross-sectional and retrospective multicentre study in two hospitals in Buenos Aires from June 2008 to June 2014. Inclusion criteria: medullary syndrome associated with spinal cord lesion extending for 3 or more contiguous spinal segments in magnetic resonance imaging (MRI). Clinical, radiological, and biochemical data were collected and subjects were rated on the Winner–Hughes Functional Disability Scale (WHFDS) at 3 months.
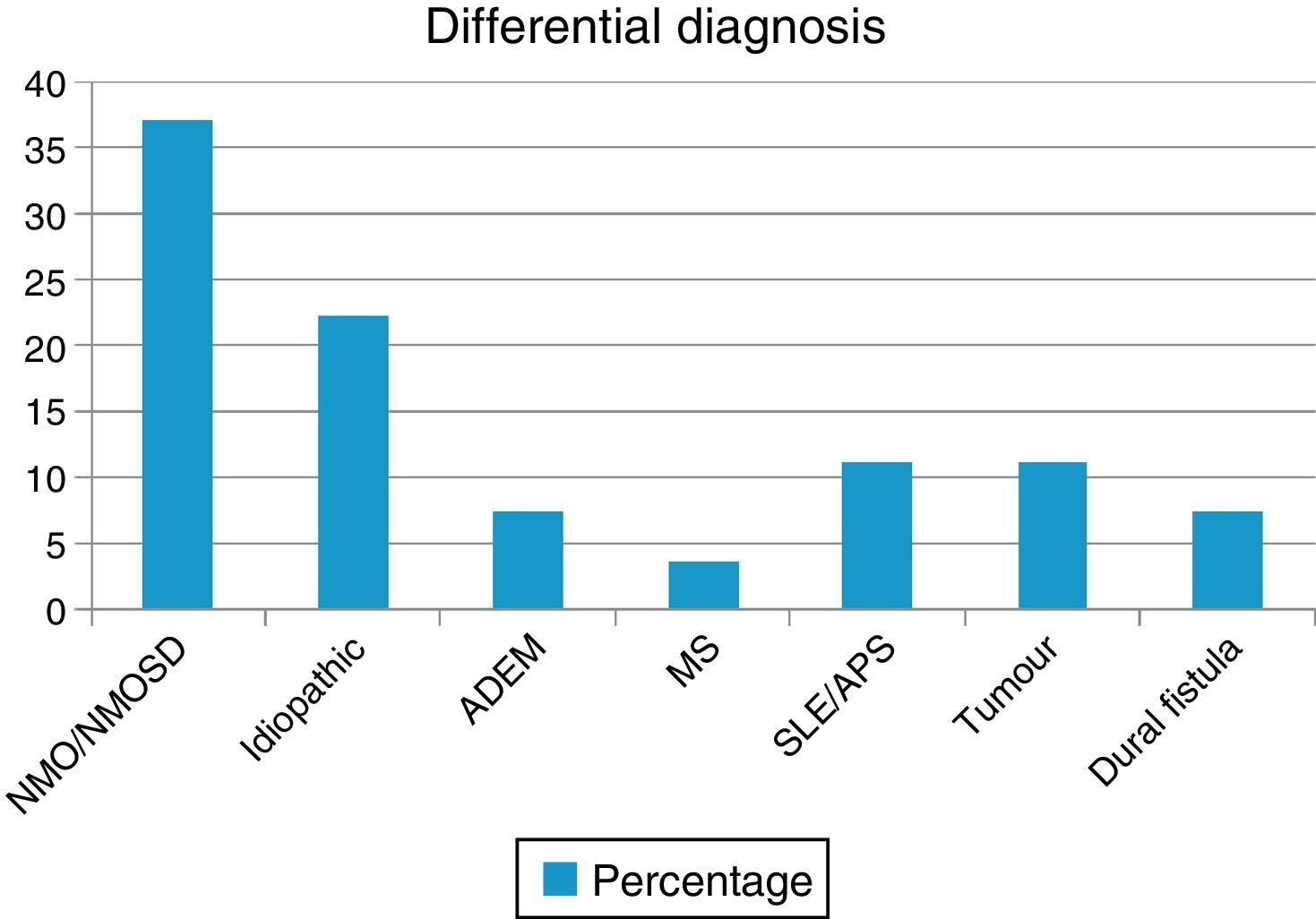
ResultsWe evaluated 27 patients, 74% of whom were women; mean age was 35.22 years. The NMO-IgG antibody test was performed in 66.6% and oligoclonal band testing in 71%. NMO-IgG seropositivity was found exclusively in NMOSD patients (75%). Brain MRI was normal in 59.2% and revealed a mean of 7.9 affected spinal segments. Differential diagnoses revealed NMOSD (37%), idiopathic LETM (22.2%), lupus (11.1%), tumour (11.1%), dural fistula (7.4%), acute disseminated encephalomyelitis (7.4%), and a single case of multiple sclerosis (3.7%). Patients with lesions to ≥7 spinal segments showed poor recovery at 3 months (P<.001); these cases were associated with neoplastic, vascular, idiopathic, and lupus-related aetiologies.
ConclusionsThe most frequent causes of LETM in our cohort were NMOSD followed by idiopathic cases. Neoplastic, vascular, lupus-related, and idiopathic LETM may constitute a critical group with a distinct prognosis and other treatment needs.
Las mielitis longitudinales extensas (LETM) fueron clásicamente relacionadas con los trastornos del espectro de la neuromielitis óptica (NMOSD) tanto definidas como limitadas. Sin embargo, los diagnósticos diferenciales incluyen una amplia gama de etiologías.
ObjetivoComunicar los diagnósticos diferenciales y el pronóstico de LETM observados en un grupo de pacientes en Buenos Aires, Argentina.
Pacientes y métodosEstudio multicéntrico retrospectivo transversal realizado en 2 hospitales de Buenos Aires desde junio del 2008 hasta junio del 2014. Criterios de inclusión: síndrome medular asociado a una lesión en la médula espinal con una extensión de 3 o más segmentos vertebrales contiguos en la resonancia magnética (RM). Datos bioquímicos, radiológicos y clínicos fueron evaluados. Asimismo, se aplicó la escala de discapacidad funcional de Winer-Hughes (WHFDS) a los 3 meses.
ResultadosSe evaluó a 27 pacientes, el 74% mujeres, edad (media): 35,22 años. NMO-IgG se realizó en el 66,6% y las bandas oligoclonales en el 71%. NMO-IgG se observó exclusivamente en pacientes con NMOSD (75%). La RM de encéfalo fue normal en el 59,2% y la media de segmentos afectados en RM espinal fue 7,9. Los diagnósticos diferenciales encontrados fueron: NMOSD (37%), idiopática (22,2%), lupus (11,1%), tumores (11,1%), fístula dural (7,4%), encefalomielitis diseminada aguda (7,4%) y esclerosis múltiple (3,7%). Los pacientes con ≥ 7 segmentos afectados tenían peor WHFDS (p < 0,001) y se asoció a etiología tumoral, vascular, lupus e idiopática.
ConclusionesEn nuestra cohorte, NMOSD seguidos por idiopática, fueron las causas más frecuentes de LETM. Las LETM tumorales, vasculares, lupus e idiopáticas pueden representar un grupo crítico con diferente pronóstico y tratamiento.
Longitudinally extensive transverse myelitis (LETM) is defined as spinal cord lesions affecting at least 3 spinal cord segments and resulting in hyperintensities on sagittal T2-weighted magnetic resonance imaging (MRI) sequences.1–3 Though rare, LETM has severe clinical consequences. A systematic evaluation to establish the aetiological diagnosis and assign early treatment are therefore essential, not only to improve short- and long-term functional outcomes but also to prevent further inflammatory-demyelinating attacks to the spinal cord and/or central nervous system.3–5 At present, LETM is mainly associated with neuromyelitis optica (NMO) and neuromyelitis optica spectrum disorders (NMOSD).1–6 However, differential diagnosis includes a number of conditions: non-NMO autoimmune inflammatory disorders (acute disseminated encephalomyelitis [ADEM], multiple sclerosis [MS], etc.), systemic autoimmune diseases (systemic lupus erythematosus [SLE], Sjögren syndrome, sarcoidosis, etc.), infections and parainfections (viral, bacterial, parasitic), paraneoplastic disorders (mainly secondary to CRMP5 antibodies), neoplasms (astrocytoma, ependymoma, spinal cord metastases, etc.), metabolic disorders (vitamin B12 or copper deficiency), and vascular diseases (stroke, dural fistulas), among others.3 In 2002, the Transverse Myelitis Consortium Working Group proposed a set of diagnostic criteria for idiopathic myelitis for those cases in which aetiology could not be established.1,2
Given the scarcity of data from our region, our purpose is to evaluate aetiological diagnoses, clinical and paraclinical characteristics, and functional outcomes at 3 months in a cohort of patients with LETM in Buenos Aires, Argentina.
Patients and methodsWe conducted a multicentre cross-sectional retrospective descriptive study of patients examined in 2 public hospitals in Buenos Aires (Hospital Carlos G. Durand and Hospital Teodoro Álvarez) between June 2008 and May 2014 by reviewing their clinical histories. We included all patients meeting the inclusion criteria (Table 1) and evaluated their demographic, clinical, paraclinical, radiological, and aetiological characteristics and functional progression at 3 months assessed on the Winner–Hughes Functional Disability Scale (WHFDS).7 Demographic data included sex and age at onset. We also checked for any motor, sensory, or autonomic disorders at the time of the neurological examination. Immunological studies, coagulation tests, and blood tests to screen for infection were conducted when there was clinical suspicion of immunological, coagulation, or infectious diseases, according to the validated international criteria for each aetiology.1,4,8–15 Likewise, serum aquaporin-4 (AQP4) antibodies (NMO-IgG) were determined by indirect immunofluorescence (confocal microscopy) on monkey cerebellum sections. Oligoclonal bands (OCB) in cerebrospinal fluid (CSF) were analysed using isoelectric focusing. We only ran specific PCR tests in cases of clinical and epidemiological suspicion. We conducted brain and spinal cord MRI studies with a 1.5-Tesla scanner. All patients underwent contrast and non-contrast MRI scans of the brain and cervical and thoracic spinal cord; MRI scans of the lumbar spinal cord were conducted when the clinician in charge deemed it necessary. Brain MRI findings were classified into (a) non-specific white matter lesions, (b) CNS lesions meeting criteria (Swanton et al.,12,13 Montalbán et al.14) for dissemination in space and time (2010 revised McDonald criteria11), or (c) typical lesions of NMO (hypothalamus, thalamus, brainstem, or cerebellum). All patients were monitored for at least 3 months; they underwent periodic neurological examinations and were assessed on the WHFDS at 90 days (0: healthy or complete recovery; 1: minor signs or symptoms which do not limit physical activity; 2: able to walk 5metres without assistance/walker/cane but unable to do daily living activities (manual work, housework, shopping); 3: able to walk 5 metres with assistance/walker/cane; 4: bedridden or wheelchair-bound; 5: needing mechanical ventilation for at least several hours during the day or at night-time; and 6: death). Patients were classified into 2 groups according to WHFDS scores: patients scoring ≤2 and patients scoring ≥3. We subsequently analysed the potential correlations between the number of affected spinal cord segments and the need for assisted walking devices (WHFDS scores ≤2 vs ≥3).
Inclusion criteria.
| Age at onset: >18 years |
| Spinal cord syndrome diagnosed by a neurologist |
| Presenting with motor/sensory impairment or sphincter dysfunction attributable to a spinal cord lesion affecting ≥3 segments and visible on MR images |
| Maximum progression time: 21 days |
| Extra-axial aetiology ruled out by imaging study |
| Spinal cord trauma ruled out |
| Gadolinium-enhanced brain and spinal cord 1.5-Tesla MRI scan (of at least the cervical and dorsal segments) |
| Minimum follow-up time with WHFDS: 3 months |
Results are expressed as proportions, percentages, and measures of central tendency (means±SD), and as measures of association (OR, 95% CI). We used Fisher's exact test to analyse statistical significance, which was set at P<.05. Statistical analysis was performed using Epi Info 7 statistical software.
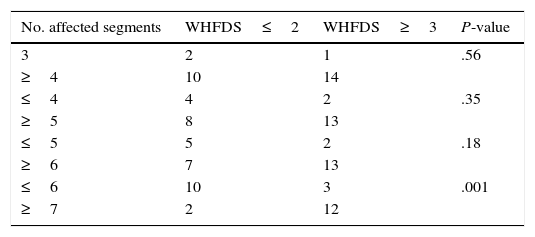
ResultsOur sample included 27 patients (N=27) with a diagnosis of LETM; 74% of our patients were women (20/27). Mean age was 38.22±16 years. All patients had sensory disorders (either superficial or deep) in at least one limb and a superficial sensory level was observed in 92.59% (25/27). The most frequently affected segments were thoracic (20/25), followed by cervical (4/25) and lumbar segments (1/25). All patients displayed motor dysfunction, which was more frequent in the lower limbs (52%). Twenty-five patients experienced sphincter dysfunction, mainly in the form of bladder dysfunction (urinary urgency, acute urinary retention). CSF oligoclonal banding was performed in 19/27 patients; results were positive in 2 cases only (10.52%). AQP4 antibodies were analysed in 18 patients within our sample; results were positive in 6 of them (33.33%). Sixteen patients (59.25%) displayed no abnormalities on brain MR images, whereas 6 (22.22%) displayed non-specific hyperintense lesions on T2-weighted and FLAIR sequences, 3 (14.81%) met the criteria for MS/ADEM, and one patient exhibited lesions compatible with NMO (areas of high AQP4 expression). Spinal cord lesions were most frequently observed in cervical-thoracic segments (13/27); the mean number of affected segments was 7.92±4.2 and the median was 7.5. According to the aetiological analysis, 10 patients (37.04%) had NMOSD (8 met the criteria for definite NMO and 2 had isolated LETM and tested positive for serum NMO-IgG [limited NMOSD]), 6 (22.22%) had idiopathic LETM, 3 (11.1%) had SLE (one also met criteria for antiphospholipid syndrome), 3 had tumours (2 with astrocytoma and one with spinal cord metastasis), 2 (7.4%) had vascular disorders (dural fistula in both cases), 2 had ADEM, and one (3.7%) had MS (Fig. 1). All patients completed the WHFDS at 3 months: 44.4% of the patients (12/27) were wheelchair-bound and 7.40% (2/27) required assistance for walking. One patient scored 6, dying at 3 months due to spinal cord metastasis secondary to lung cancer. Broken down by need for assistance for walking at least 5 metres, 15 patients scored ≥3 and the remaining 12 patients scored ≤2. We analysed the association between the number of affected spinal segments and WHFDS scores (≤2 vs ≥3) and found that patients with ≥7 affected segments had a poorer functional prognosis (95% CI, OR 20.0; P<.001) (Tables 2 and 3).

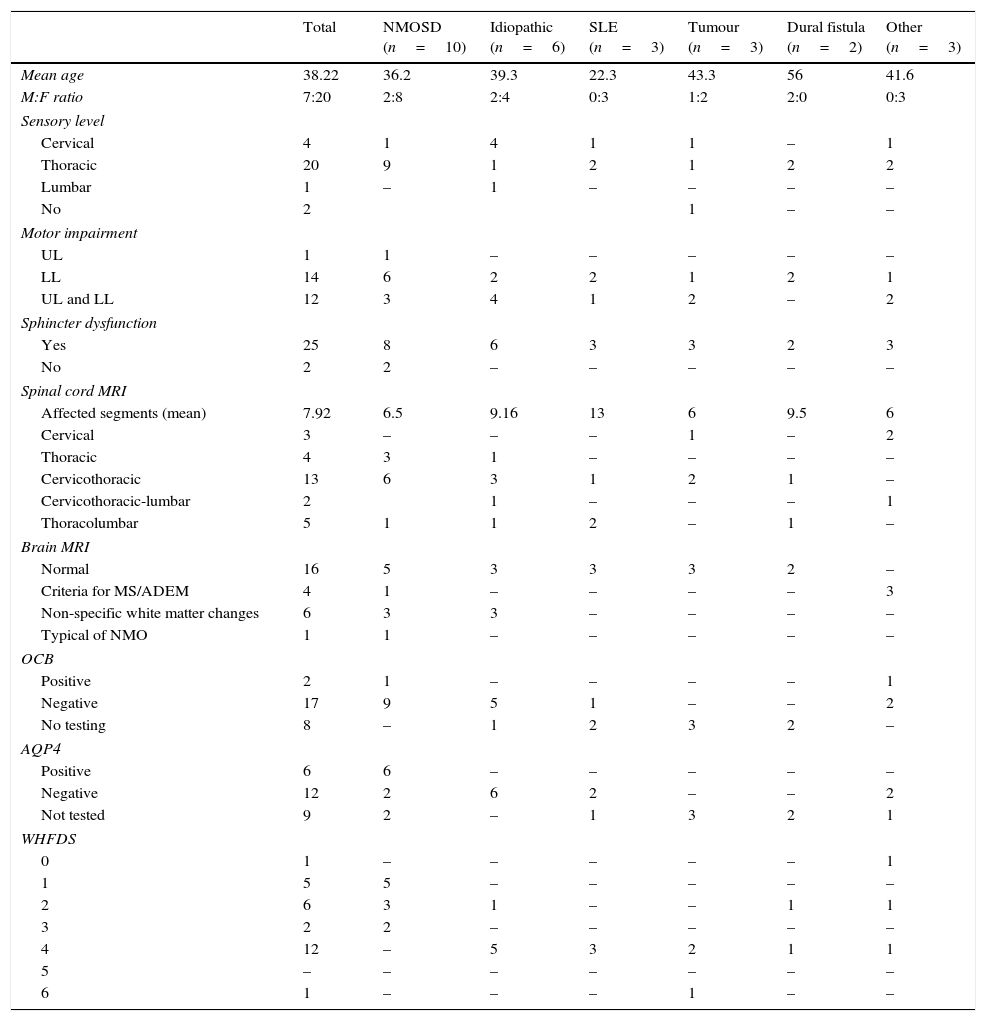
Demographic, clinical, and paraclinical characteristics; neuroimaging findings; and prognosis in our series.
| Total | NMOSD (n=10) | Idiopathic (n=6) | SLE (n=3) | Tumour (n=3) | Dural fistula (n=2) | Other (n=3) | |
|---|---|---|---|---|---|---|---|
| Mean age | 38.22 | 36.2 | 39.3 | 22.3 | 43.3 | 56 | 41.6 |
| M:F ratio | 7:20 | 2:8 | 2:4 | 0:3 | 1:2 | 2:0 | 0:3 |
| Sensory level | |||||||
| Cervical | 4 | 1 | 4 | 1 | 1 | – | 1 |
| Thoracic | 20 | 9 | 1 | 2 | 1 | 2 | 2 |
| Lumbar | 1 | – | 1 | – | – | – | – |
| No | 2 | 1 | – | – | |||
| Motor impairment | |||||||
| UL | 1 | 1 | – | – | – | – | – |
| LL | 14 | 6 | 2 | 2 | 1 | 2 | 1 |
| UL and LL | 12 | 3 | 4 | 1 | 2 | – | 2 |
| Sphincter dysfunction | |||||||
| Yes | 25 | 8 | 6 | 3 | 3 | 2 | 3 |
| No | 2 | 2 | – | – | – | – | – |
| Spinal cord MRI | |||||||
| Affected segments (mean) | 7.92 | 6.5 | 9.16 | 13 | 6 | 9.5 | 6 |
| Cervical | 3 | – | – | – | 1 | – | 2 |
| Thoracic | 4 | 3 | 1 | – | – | – | – |
| Cervicothoracic | 13 | 6 | 3 | 1 | 2 | 1 | – |
| Cervicothoracic-lumbar | 2 | 1 | – | – | – | 1 | |
| Thoracolumbar | 5 | 1 | 1 | 2 | – | 1 | – |
| Brain MRI | |||||||
| Normal | 16 | 5 | 3 | 3 | 3 | 2 | – |
| Criteria for MS/ADEM | 4 | 1 | – | – | – | – | 3 |
| Non-specific white matter changes | 6 | 3 | 3 | – | – | – | – |
| Typical of NMO | 1 | 1 | – | – | – | – | – |
| OCB | |||||||
| Positive | 2 | 1 | – | – | – | – | 1 |
| Negative | 17 | 9 | 5 | 1 | – | – | 2 |
| No testing | 8 | – | 1 | 2 | 3 | 2 | – |
| AQP4 | |||||||
| Positive | 6 | 6 | – | – | – | – | – |
| Negative | 12 | 2 | 6 | 2 | – | – | 2 |
| Not tested | 9 | 2 | – | 1 | 3 | 2 | 1 |
| WHFDS | |||||||
| 0 | 1 | – | – | – | – | – | 1 |
| 1 | 5 | 5 | – | – | – | – | – |
| 2 | 6 | 3 | 1 | – | – | 1 | 1 |
| 3 | 2 | 2 | – | – | – | – | – |
| 4 | 12 | – | 5 | 3 | 2 | 1 | 1 |
| 5 | – | – | – | – | – | – | – |
| 6 | 1 | – | – | – | 1 | – | – |
ADEM: acute disseminated encephalomyelitis; AQP4: aquaporin-4 antibodies; OCB: oligoclonal bands; MS: multiple sclerosis; F: female; M: male; LL: lower limbs; UL: upper limbs; NMO: neuromyelitis optica; MRI: magnetic resonance imaging.
Our retrospective series included 27 patients diagnosed with LETM (Fig. 2). In line with the literature, LETM was more frequent in women; mean age in our sample was 38.22 years. Interestingly, patients with a vascular aetiology were approximately 10 years older (mean: 56 years) than the rest of the patients; 2 previous studies found that female sex and age under 40 years were more strongly associated with inflammatory myelopathies than with vascular diseases.16,17 The most frequent aetiology in our series was NMO (36%), followed by idiopathic LETM (22.2%); only one patient had MS (3.7%) (Table 2). In a study conducted in the United Kingdom18 and including 76 patients with LETM (44 AQP4-Ab–positive and 32 AQP4-Ab–negative), 53 patients were diagnosed with NMO (44 AQP4-Ab–positive, 4 MOG-Ab–positive, and 5 seronegative), 5 had ADEM, 5 had idiopathic LETM, 4 had infectious causes (1 tuberculosis, 1 mycoplasma, 1 Epstein–Barr virus, 1 West Nile virus), 3 had MS, 2 had LETM associated with MOG-Ab–positivity, 2 had other inflammatory diseases, 1 had a paraneoplastic disease, and 1 had vascular disease. Likewise, in a study conducted in Brazil,19 NMO was present in 46.8% of the sample, recurrent LETM in 14%, isolated LETM in 16%, AV fistulas in 4.4%, and rheumatic disease in 4.4%. In that study, 44% of the recurrent cases and 20% of the isolated cases were positive for AQP4. The remaining patients were negative for NMO-IgG.15 These findings agree with the data obtained in our study.20 However, one multicentre retrospective study21 conducted in hospitals in Spain and France and including 72 patients found that 22 had idiopathic LETM, 18 MS, 11 parainfectious diseases, 9 systemic disorders, 3 NMO, 3 vascular diseases, 2 ADEM, 2 dural fistulas, and 2 tumours. Another study conducted in West Australia22 evaluated 26 patients with LETM and identified LETM as the initial presentation in 13 patients (1 monophasic, 4 MS, 3 NMO, and 5 recurrent), whereas the remaining 13 patients (9 MS and 4 NMO) developed LETM during follow-up (1-27 years). The aetiological heterogeneity of LETM across regions and populations is noteworthy. Both studies report higher rates of MS and lower rates of NMO than does our study.
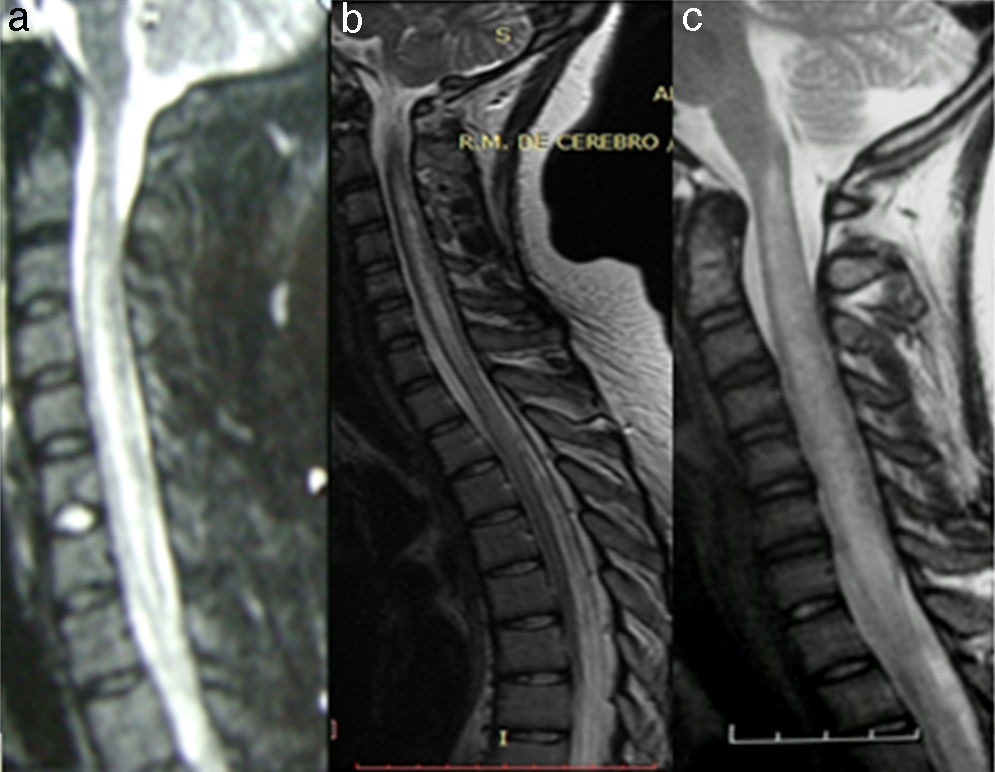
 Extensive hyperintense lesion in a patient with ADEM. (b) Extensive hyperintense lesion in an NMO seropositive patient. (c) Extensive hyperintense lesion in a patient with SLE.")
Although clinical manifestations are widely known; however, they are not useful for aetiological diagnosis according to several series. Regarding complementary laboratory tests, 70.37% of our patients underwent CSF oligoclonal banding; results were positive for 10.52% (2/19). One patient with NMO and the only patient with MS were found to be OCB-positive. Several studies have shown the relevance of oligoclonal banding for diagnosing MS and predicting outcomes and conversion to clinically definite MS in patients with a first episode of atypical myelitis.23,24 Around 15% to 30% of the patients are positive for NMO-IgG4; these rates are similar to our own. In 2004, Lennon et al.25 found that an antibody called NMO-IgG (AQP4) was sensitive and specific for distinguishing NMO from MS. In our series, AQP4 antibodies were determined in 66.66% of our sample and were found to be present in 75% of the patients with NMO (2 patients were not tested for AQP4 antibodies since the test was not available and they had already met the diagnostic criteria for definite NMO). Testing for these antibodies in patients with a first episode of LETM is essential not only to diagnose NMOSD (limited forms) but also to predict outcomes, as shown by the preliminary study by Weinshenker et al.26; these researchers observed that 56% of seropositive patients with LETM developed definite NMO after 2 years. This underscores the need for early immunosuppressant treatment to avoid further episodes of myelopathy and/or optic neuritis.26–28 We stress the lack of availability of specific tests for detecting NMO-IgG and monitoring these antibodies in the long term; this was the reason why we were unable to determine the real percentage of patients with NMOSD-related LETM. However, all patients with idiopathic LETM underwent NMO-IgG testing and were found to be seronegative.28,29 After the initial clinical assessment, it is difficult to determine the aetiology. Likewise, in a study conducted in France30 and including 170 patients, the initial assessment was unable to identify a definite cause of myelitis in 101 patients (59.4%), 55 of whom were subsequently diagnosed with inflammatory/demyelinating myelitis at the end of follow-up; only 49 of the total (28.8%) received a definitive aetiological diagnosis of either idiopathic myelitis, MS, or NMO. In line with the literature, 59.2% of our patients showed no abnormalities in brain MR images. Pathological results were found in patients with ADEM and MS; 30% of patients with NMO and 50% of those with idiopathic LETM displayed non-specific lesions. Around 7% to 9% of the patients with NMO/NMOSD display lesions in regions of high AQP4 expression (10% in our series; 1/10).31–34 Regarding patients’ functional status at 3 months (WHFDS), 44.4% of our patients were wheelchair-bound. A high percentage of patients with tumours, vascular diseases, systemic diseases (SLE), and idiopathic LETM were wheelchair-bound (WHFDS 4). We found a correlation between presence of ≥7 affected segments and need for assistance when walking (P<.001). According to a recent study of 23 patients, the number of spinal cord segments affected by LETM was not correlated with poorer prognosis, but it was found to be associated with the risk of recurrence.7 Our study has several limitations inherent to its retrospective design. It does not provide data on NMO-IgG and OCB for all patients, and lacks long-term follow-up and a comparison between WHFDS scores at baseline and at 3 months. Nevertheless, it does provide an overall idea of the clinical, paraclinical, aetiological, and demographic features of LETM in a sample of the general population drawn from 2 public hospitals in Buenos Aires. Only a few clinical studies of LETM have been carried out in Argentina. Having a deeper knowledge of our population is essential if we are to compare our results with results from studies conducted in other countries. Furthermore, given the implications for treatment, performing an exhaustive aetiological assessment is extremely important so as to distinguish between the multiple causes underlying LETM.
ConclusionsThe most frequent aetiologies in our sample were NMOSD followed by idiopathic LETM. NMO-IgG antibodies play a crucial role in differential diagnosis. In our sample, clinical characteristics were not useful for aetiological diagnosis. Functional prognosis in patients with ≥7 affected segments was poor. On the other hand, half of the patients with NMOSD had a good functional prognosis. We should maintain a high level of suspicion for multiple potential aetiologies, especially tumours, vascular diseases, SLE, and other unknown causes; these cases may represent a subgroup of LETM with different treatment needs and prognosis.
FundingThis study has received no funding of any kind.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Carnero Contentti E, Hryb JP, Leguizamón F, Di Pace JL, Celso J, Knorre E, et al. Diagnósticos diferenciales y pronóstico de las mielitis longitudinales extensas en Buenos Aires, Argentina. Neurología. 2017;32:99–105.
