



En el mundo, alrededor de 50 millones de personas padecen demencia; la forma más común es la enfermedad de Alzheimer (EA), que representa el 60-70% de los casos. Dada su alta incidencia, se hace imperativo diseñar estudios que permitan ampliar el conocimiento sobre su aparición y desarrollo, para proponer diagnósticos tempranos y/o posibles tratamientos. Una de las estrategias metodológicas que se han desarrollado son los modelos transgénicos murinos para el estudio de los factores involucrados en su etiología, y entre ellos, el estrés oxidativo y la respuesta inmune.
DesarrolloSe realizó una búsqueda de artículos originales y revisiones en PubMed, Scopus y Google Scholar (2013-2019). En esta revisión abordamos dos factores que han sido estudiados de forma independiente: el estrés oxidativo y la respuesta inmune en modelos transgénicos para la EA, y se discute la relación que existe entre ellos y que impacta en la pérdida de la plasticidad sináptica y estructural, produciendo como efecto final el deterioro cognitivo.
ConclusiónEsta revisión describe posibles mecanismos en donde participan el estrés oxidativo y la respuesta inmune sobre los efectos moleculares, celulares y conductuales en la EA, observando una estrecha relación entre estos elementos que conducen hacia el deterioro cognitivo.
Worldwide, approximately 50 million people have dementia, with Alzheimer disease (AD) being the most common type, accounting for 60%-70% of cases. Given its high incidence, it is imperative to design studies to expand our knowledge about its onset and development, and to develop early diagnosis strategies and/or possible treatments. One methodological strategy is the use of transgenic mouse models for the study of the factors involved in AD aetiology, which include oxidative stress and the immune response.
DevelopmentWe searched the PubMed, Scopus, and Google Scholar databases for original articles and reviews published between 2013 and 2019. In this review, we address two factors that have been studied independently, oxidative stress and the immune response, in transgenic models of AD, and discuss the relationship between these factors and their impact on the loss of synaptic and structural plasticity, resulting in cognitive impairment.
ConclusionThis review describes possible mechanisms by which oxidative stress and the immune response participate in the molecular, cellular, and behavioural effects of AD, observing a close relationship between these factors, which lead to cognitive impairment.
La demencia es un síndrome, usualmente de naturaleza crónica o progresiva, que causa deterioro cognitivo. En el mundo, alrededor de 50 millones de personas padecen demencia, y su incidencia aumenta cada año. Existen diferentes formas de demencia; la más común es la enfermedad de Alzheimer (EA), que representa el 60-70% de los casos1.
La EA es un trastorno neurodegenerativo en el cual se observan placas neuríticas y difusas de la proteína beta amiloide (βA) y marañas de la proteína tau hiperfosforilada en el tejido cerebral2.
La EA es una patología neurodegenerativa multifactorial, y a medida que aumentan el número de estudios en el área se suman nuevos elementos que son importantes para la aparición y el desarrollo de la enfermedad. Así mismo, esto ha hecho más complejo el estudio y el diseño de posibles tratamientos que puedan incidir desde la fase asintomática de la EA.
El uso de los modelos murinos transgénicos que desarrollan la EA procura una gran ventaja en la obtención de información concerniente a la aparición, el desarrollo, el diagnóstico temprano y tardío de la patología, así como posibles estrategias para evitar la aparición de esta enfermedad, controlar la sintomatología o incluso diseñar tratamientos para la eliminación de este tipo de demencia. Sin embargo, en los modelos existentes no se ha logrado replicar completamente toda la sintomatología observada en la EA en humanos. En esta revisión, basada en el estudio de los modelos transgénicos, nos referiremos a la EA como la enfermedad tipo Alzheimer.
Actualmente existen más de 160 modelos transgénicos para la EA que varían en función del gen que es modificado (mutado) o del número de proteínas alteradas (sobreexpresadas). Los modelos más utilizados para el estudio de la EA se muestran en la tabla 1; algunos son doble o triple-transgénicos, basados en el metabolismo de la proteína precursora del amiloide (APP), de la presenilina (PSN), o de ambas. Otros se basan en la proteína tau y los triple-transgénicos PSN/APP/tau (Manzano et al., 2009). Hay líneas que presentan alteración en la apolipoproteína (ApoE). En estos modelos se estudian de manera particular el efecto de los oligómeros de βA (especies tóxicas solubles).
Número de artículos publicados en los últimos 5 años (2019, 2018, 2017, 2016, 2015, respectivamente) en la base de datos PubMed de los estudios de la EA en cinco modelos murinos transgénicos (3xTg-AD, 5xFAD, Tg2576, APP/PS1 y rTg4510). Cuatro de estos modelos basados en el metabolismo de la proteína precursora del amiloide (APP), tres modelos con mutación en la presenilina 1 (PSN1) y solo dos modelos con una mutación en la proteína tau (tau)
| Modelo | Genes mutados o proteínas sobreexpresadas | Número de artículos publicados | ||||
|---|---|---|---|---|---|---|
| 2019 | 2018 | 2017 | 2016 | 2015 | ||
| 3xTg-AD | APP, PSEN1, tau | 10 | 47 | 44 | 39 | 49 |
| 5xFAD | APP, PSEN1 | 12 | 67 | 62 | 50 | 56 |
| Tg2576 | APP | 8 | 20 | 41 | 42 | 58 |
| APP/PS1 | APP, PSEN1 | 2 | 26 | 19 | 29 | 16 |
| rTg4510 | Tau | 3 | 12 | 8 | 9 | 6 |
En esta revisión enfocamos nuestra atención en la participación que tienen el estrés oxidativo y la respuesta inmune sobre la EA en diferentes modelos transgénicos que desarrollan la patología. A su vez, se revisa cómo se afectan la plasticidad sináptica y la cognición durante el desarrollo de la EA.
El estrés oxidativoEl estrés oxidativo ha sido ampliamente vinculado a enfermedades neurodegenerativas3-5, entre las que se encuentra la EA6-9. El estrés oxidativo y la oligomerización de la proteína βA preceden el inicio de la demencia clínica en la EA10. Estudios realizados a varias cepas de ratones que sirven como modelos transgénicos de la EA presentan aumentados los indicadores de estrés oxidativo11.
El cerebro es particularmente vulnerable al daño por estrés oxidativo, debido a su alto consumo de oxígeno, a los altos niveles de ácidos grasos poliinsaturados, a los niveles bajos de antioxidantes y a la presencia de una concentración relativamente alta de iones metálicos de transición redox (para revisiones, ver Moreira et al.12 y Butterfield13).
El diseño de ratones transgénicos, que expresan diferentes mutaciones relacionadas con la EA, ha permitido estudiar las implicaciones del daño por estrés oxidativo en esta enfermedad. Algo importante de mencionar es que los marcadores de estrés oxidativo (la gran mayoría productos de la lipoperoxidación) son observables con antelación a la aparición de las placas de βA y de los efectos cognitivos de la EA, permitiendo así el estudio de estos marcadores para un diagnóstico temprano de dicha enfermedad7,14.
Entre los daños más comunes causados por el estrés oxidativo se encuentran la lipoperoxidación, la cual da lugar a la formación de F2-isoprostanos (F2-IsoPs), F4-neuroprostanos (F4-NeuroPs) malondialdehído (MDA) y 4-hidroxi-2-nonenal (HNE); dichas moléculas son usadas como biomarcadores de estrés oxidativo in vivo14-17. También se ha observado oxidación de proteínas, cuyo principal marcador es la carbonilación proteica18,19. Por último se encuentra la oxidación de bases nitrogenadas, siendo la 8-oxo-2’-desoxiguanosina (8-OHdG) y la 8-hidroxiguanosina (8-OHG) los principales marcadores de daño oxidativo en el ADN y el ARN, respectivamente20-22.
Por ejemplo, la mutación APP sueca encontrada en Tg2576 induce un aumento de F2-IsoPs en orina, plasma y tejido cerebral a los 8meses de edad, es decir, entre 2 y 4meses antes de la aparición de las placas βA y del deterioro en la memoria espacial. Además, dicho aumento se hace más pronunciado a los 12 y 18meses de edad, lo que correlaciona con el inicio y la progresión de la acumulación de las placas βA23. Por otro lado, también se ha observado un incremento en la detección de HNE inmunorreactivo en neuritas distróficas adyacentes a las placas βA24. En cuanto a la oxidación directa de proteínas, se ha observado un incremento de carbonilos proteicos en el hipocampo de estos ratones transgénicos25-27.
La cepa Thy1-APP751SL presenta una reducción de la actividad de la enzima antioxidante Cu/Zn-SOD y un incremento de la lipoperoxidación detectado por medio del marcador HNE. Esta cepa se caracteriza por una sobreexpresión de la isoforma 751 de la APP humana (APP751) y contiene las mutaciones londinense y sueca, lo que induce una alta expresión de APP, altos niveles de βA y el inicio de la formación de placas a una edad de 6meses28.
La cepa PDGF-APP695SDL, que contiene la isoforma APP695, con las mutaciones sueca, alemana y londinense, presenta niveles más bajos de formación de detección de placas βA, los cuales son observables hasta una edad de 18meses28. Esta cepa no presenta la reducción de la actividad de Cu/Zn-SOD ni el incremento de la lipoperoxidación, lo que sugiere que la reducción de la actividad antioxidante que conduce a un daño oxidativo está relacionada con el procesamiento β-amiloidogénico de APP29.
Con respecto a los transgénicos que contienen el gen PSEN1, se ha demostrado que este gen induce la acumulación temprana de βA42 en las placas seniles30. Ratones que expresan la PS-1 mutante humana presentan un aumento en los marcadores de estrés oxidativo, carbonilación proteica y HNE31. Además, ratones doble-transgénicos para APP/PSEN1 presentan un aumento mayor de estos marcadores, así como un incremento de F2-IsoPs31,32.
Los ratones transgénicos P301S de la línea PS19 son un modelo ampliamente usado en el estudio de la taupatía en la EA, y expresan la proteína tau humana mutante P301S. Este transgénico presenta una alta acumulación de hierro en la corteza y en el hipocampo a los 7meses y medio de edad33. La acumulación de hierro se ha vinculado a la agregación de la proteína tau hiperfosforilada y a sus efectos en el desbalance REDOX34. Además, también se ha observado en estos mismos ratones una disminución en la expresión de GPx4, xCT y SOD1, enzimas antioxidantes importantes, así como una disminución en la actividad de SOD33. También se ha observado que ratones transgénicos que expresan la proteína tau humana presentan un incremento de la carbonilación proteica en la corteza cerebral, así como un incremento de la 8-oxo-2’-desoxiguanosina (8-OHdG), un marcador de daño oxidativo en ácidos nucleicos35.
Se ha observado que en ratones conocidos como triple-transgénicos (3xTg-AD), debido a que expresan APP con la mutación sueca, presenilina humana PSEN1 y la TAU humana mutante MAPT P301L36, se presenta una reducción de los niveles de los antioxidantes glutatión reducido y vitaminaE, además de un incremento en la peroxidación lipídica. También se ha observado una mayor actividad de las enzimas antioxidantes glutatión peroxidasa y superóxido dismutasa. Estas alteraciones son observables durante el período de oligomerización del βA y antes de la aparición de las placas βA y los ovillos neurofibrilares, es decir, el estrés oxidativo se produce de forma temprana durante el desarrollo de la enfermedad37.
También se ha observado en un trabajo reciente que el hipocampo de 3xTg-AD presenta el doble de acumulación de especies reactivas de oxígeno y de nitrógeno, lo que explica la acumulación de marcadores de lipoperoxidación HNE, MDA y 8-iso PGF2α38. En cuanto a daños a nivel proteico, se observó un incremento en la carbonilación proteica y la nitración de tirosinas19,38. Por último, también se demostró un incremento en los niveles de 8-OHdG y de 8-OHG, marcadores de daño oxidativo en el ADN y ARN, respectivamente38.
La respuesta inmune y la microglíaLa participación de la inflamación en la EA fue inicialmente descrita por Oskar Fischer casi simultáneamente con Alois Alzheimer, pero el estudio de la respuesta inmune en el sistema nervioso central en la EA fue escasa durante mucho tiempo; últimamente ha cobrado importancia39, ya que la evidencia emergente sugiere que la inflamación tiene un papel causal en la EA, y su contribución en el desarrollo de la patología puede ser tanro o más importante que tau y βA40.
En investigaciones realizadas con la asociación del genoma completo (GWAS por sus siglas en inglés) se ha encontrado una relevante participación de la respuesta inmune de la microglía y la EA41. La mayoría de los genes que se han encontrado como factores de riesgo están presentes en la microglía, como es el caso de TREM2 y APOE41,42.
Se ha sugerido la participación de una respuesta proinflamatoria por parte de la microglía en la EA, ya que se ha encontrado que la morfología de la microglía asociada a las placas de βA y/o a las marañas de tau en pacientes post mortem con la enfermedad es distinta a la observada en eventos proinflamatorios como la sepsis; a esta microglía con morfología distinta se la denomina microglía distrófica o microglía senescente43. Debido a esto, se ha propuesto que quizás este tipo de microglía es la que está relacionada con la disfunción sináptica y la neurodegeneración observada en la patología, y no la proinflamatoria, como se pensaba anteriormente43. En el modelo transgénico 5xFAD también se ha observado un fenotipo de microglía distinto al proinflamatorio, al cual se le ha denominado microglía asociada a enfermedad (disease-associated microglia [DAM]) y que se caracteriza por tener una sobreexpresión de TREM2 y APOE44. De igual forma, en el modelo transgénico APP-PS1 se ha reportado un fenotipo de microglía distinto al proinflamatorio cerca de las placas de βA, al que se le ha denominado MGnD (por sus siglas en inglés, molecules signature of disease-associated microglia). Estos fenotipos de la microglía observados en los tres modelos transgénicos para la EA tienen sobreexpresados los genes TREM2 y APOE45 asociados como factor de riesgo para desarrollar la EA46. Es importante mencionar que la expresión de estos genes se ve inhibida en el fenotipo proinflamatorio inducido con lipopolisacárido (LPS) o el interferón gamma (IFN-γ)45.
La microglía tiene una participación importante durante el desarrollo en la eliminación de neuronas que no pertenecen a circuitos funcionales y remodelando las sinapsis por medio de C1q y C3 de la vía del complemento47. Las sinapsis que se eliminarán inicialmente son marcadas con estas proteínas del complemento y posteriormente son reconocidas por el receptor C3 (C3R) de la microglía para su posterior eliminación47. Este proceso ocurre mediante el proceso denominado trogocitosis*, el cual solo está restringido a botones presinápticos y axones48. Este hallazgo resulta interesante, ya que la principal característica de la EA es la presencia de placas βA y de las marañas de tau; actualmente se incluye la pérdida de sinapsis y el deterioro cognitivo, así como la neurodegeneración en etapas tardías49, en la que se sugiere participación de la microglía.
En modelos amiloidogénicos como el ratón transgénico 5xFAD se ha observado disfunción sináptica, pérdida de sinapsis y muerte neuronal50; sin embargo, si se elimina la microglía se evita la pérdida de espinas dendríticas y la pérdida neuronal, sin que se observen cambios en los niveles de βA o en el tamaño de las placas βA, lo cual sugiere que el fenotipo de microglía encontrado en este modelo es el causante de la pérdida de las espinas y de la muerte neuronal42,51. Por otro lado, se ha observado que la proteína βA tiene una participación importante en la poda dendrítica, pues los fenotipos DAM y MGnD se encuentran en los estadios tardíos de la EA en los modelos murinos amiloidogénicos y no se encuentran en los ratones no transgénicos44,45. Además, los oligómeros de βA pueden activar la vía del complemento, provocando que C1q marque las sinapsis que más tarde serán reconocidas por el C3R de la microglía, provocando la eliminación de las sinapsis52.
Existe evidencia que muestra que en las placas βA se han encontrado proteínas de la vía del complemento39, de las cuales C3 y C3R en ratones no transgénicos favorecen la fagocitosis de la proteína βA por parte de la microglía53. Esto podría sugerir que la presencia de las proteínas del complemento en las placas βA serviría para marcarlas y así ser reconocidas por la microglía para su posterior eliminación.
Por otro lado, se ha observado en el modelo amiloidogénico como el APP, que el C3R en microglía suprime su capacidad de fagocitar la proteína βA54 y al eliminar C3 en el modelo APP/PS1 (APP/PS1 C3KO) se evita la pérdida de sinapsis, aunque no exista una disminución de placas βA55. Estas diferencias observadas en los modelos no transgénicos con respecto los modelos transgénicos APP/PS1 se pueden deber al fenotipo de la microglía, lo que sugiere que en el fenotipo proinflamatorio el C3 y el C3R protegen a la neurona, mientras que en los fenotipos DAM y MGnD provocan la pérdida de las sinapsis y la neurodegeneración.
Resulta interesante que los fenotipos DAM y MGnD sobreexpresan TREM2 en la proximidad de las placas βA44,45, ya que este receptor tiene preferencia por los oligómeros de la proteína βA46, los cuales pueden inducir la eliminación de sinapsis mediante la vía del complemento52.
Ahora surge la pregunta: ¿Cuál es la causa de que la microglía pierda la homeostasis y elimine sinapsis de manera descontrolada? Una de las posibles explicaciones es que el gen que codifica para el receptor1 a quimiocina (CX3CR1) está involucrado; este gen se encarga de inhibir a la microglía y es uno de los genes cuya expresión se ve disminuida en los estadios tempranos en modelos transgénicos amiloidogénicos, lo que puede causar que en estadios tardíos de la patología se sobreexprese TREM244.
Por último, se ha observado que la microglía también tiene una participación importante en el modelo transgénico MAPTP301S (cuya principal característica es el desarrollo de la taupatía), ya que al eliminar este tipo celular, así como a los astrocitos senescentes, se evita la formación de marañas de tau y la neurodegeneración56. Esto sugiere, al igual que en los modelos amiloides, que un fenotipo microglial distinto al proinflamatorio es el involucrado en la EA.
La plasticidad sináptica y la cogniciónLa plasticidad sináptica se define como el mecanismo que induce cambios en la eficiencia sináptica como resultado de la actividad neuronal de alta frecuencia derivada de la experiencia57. La plasticidad sináptica comprende una variedad amplia de modificaciones estructurales y funcionales, tales como la potenciación a largo plazo (LTP), la sinaptogénesis, el remodelamiento dendrítico y axonal, y en algunos casos se incluye la neurogénesis en el adulto58.
Algunos de estos mecanismos de plasticidad sináptica estructural implican el remodelamiento sináptico, es decir, cambios estructurales en las dendritas, específicamente en las espinas dendríticas. Las espinas dendríticas son compartimentos celulares hechos principalmente de elementos del citoesqueleto59, y además contienen en la estructura interna un complejo proteico denominado densidad postsináptica (PSD, por sus siglas en inglés); en este complejo se encuentran ancladas una gran variedad de moléculas, como es el caso de los receptores de algunos neurotransmisores, como los receptores AMPA, NMDA y kainato60. Las espinas dendríticas varían morfológicamente, lo cual permite que sean clasificadas según su forma en espinas dendríticas tipo larga, hongo y corta61. La morfología de las espinas dendríticas está relacionada con su función, es decir, las espinas tipo larga se han relacionado con procesos cognitivos como el aprendizaje, mientras que las espinas tipo hongo se han relacionado con la memoria62 y las espinas tipo corta han sido propuestas como reguladores de la excitabilidad a través de la regulación del ion calcio63.
Existen estructuras cerebrales como el hipocampo, la amígdala, el estriado y la corteza que participan activamente en el procesamiento de información asociada con el aprendizaje y la memoria; estos procesos cognoscitivos inducen cambios en la plasticidad sináptica en condiciones normales64-68, pero en pacientes o en modelos transgénicos estos procesos se ven afectados por el desarrollo de la EA. En los modelos transgénicos para la EA se ha estudiado cómo esta patología afecta la plasticidad sináptica. Estos trabajos han sido realizados en tres niveles de análisis: molecular, celular y cognitivo-conductual.
A nivel molecular, en el modelo 3xTg-AD se ha observado que en hembras de 3meses de edad se encuentra aumentada la expresión del gen GluA3 y GluA4 en el hipocampo; sin embargo, a los 10meses de edad este gen se encuentra disminuido en comparación con ratones no transgénicos69; estos resultados sugieren que el sistema de transmisión glutamatérgico se ve afectado por la patología. Así mismo, se ha observado que la expresión de la proteína Arc (activity-regulated cytoskeleton-associated protein) se modifica en el hipocampo dorsal de ratones 3xTg-AD. La proteína Arc ha sido utilizada como un marcador de actividad neuronal70, ya que su gen es de expresión inmediata temprana, y también se utiliza como marcador de plasticidad sináptica por su participación en la endocitosis de los receptores AMPA y en el escalamiento sináptico71. En ratones 3xTg-AD macho de 10meses de edad la expresión basal de la proteína Arc está aumentada en el CA3 del hipocampo; no obstante, cuando estos animales son entrenados en el laberinto acuático de Morris la expresión de la proteína Arc no cambia en comparación con su basal; además, la mayoría de estas células Arc positivas son neuronas que presentan placas βA72. Evidencia reciente sugiere que la proteína Arc juega un papel importante en la sinaptopatía de la EA73.
En otro estudio realizado en ratones 3xTg-AD macho de un mes de edad se ha observado que la fosforilación de la proteína tau está relacionada con cambios en las oscilaciones theta hipocampales e induce una reducción en la excitabilidad en neuronas que se encuentran en CA1/subículo74. En el mismo modelo también se ha reportado un deterioro en la potenciación a largo plazo (LTP) por la formación de placas βA y marañas de tau75. Otro estudio realizado en ratones 3xTg-AD de 12 a 16meses de edad se encontró un aumento en la corriente del canal de Ca+2 dependiente de voltaje (L-VGCC) en el CA1 del hipocampo, aumentando la concentración de Ca+2 en el interior de la neurona76, lo cual terminaría en una excitotoxicidad neuronal77,78.
La neurotransmisión activa múltiples cascadas de señalización específicas en función del receptor activado y de la experiencia a la que sea sometido un individuo. Algunas de estas vías de señalización inducen la expresión de genes que codifican para proteínas estructurales, que son las responsables de la re-estructuración dinámica neuronal, como es el caso de las espinas dendríticas.
En ratones transgénicos βA-PP/PS1 de 13meses de edad se observó que la EA modifica los niveles de las proteínas como ERK44, pERK44, c-Fos, mTOR y βA-PP en la corteza parietal, el CA1 y el giro dentado del hipocampo dorsal79. Esto implica que las cascadas de señalización en las que participan estas proteínas se ven afectadas, por lo que la señalización molecular río abajo se altera, incidiendo indirectamente en moléculas importantes para la plasticidad sináptica estructural como la Ca2+/calmodulina/CaMKII/CaV1.280, las cuales afectarían a proteínas de citoesqueleto como la F-actina. Esta es otra de las vías por la se puede interferir con la plasticidad sináptica estructural, ya que, como se había comentado previamente, la trogocitosis dada por la microglía también causa una pérdida de la sinapsis, lo cual podría implicar directamente una pérdida de las espinas dendríticas.
En el modelo transgénico APP/PS1 de 1-2meses de edad se ha observado que la proteína βA induce la despolimerización de la F-actina, lo cual induce una disminución en la densidad de espinas dendríticas81. De la misma forma, en los modelos transgénicos APP23 y APPswe/PS1deltaE9 de 4meses de edad se ha observado una disminución en la densidad de espinas dendríticas cerca de las placas de proteína βA, así como una disminución en las espinas dendríticas tipo hongo, las cuales se han relacionado con procesos cognitivos como la memoria82.
En los modelos para la EA se ha relacionado la disfunción sináptica con el deterioro cognitivo. En el modelo 3xTg-AD se ha observado una afectación en la ejecución de paradigmas conductuales aprendidos e innatos. En las conductas aprendidas se ha observado una disminución en el tiempo de permanencia en los brazos abiertos en el laberinto elevado, un menor índice de discriminación en la tarea de reconocimiento de objetos y un deterioro en la retención en el laberinto acuático de Morris83. En hembras 3xTg-AD de 8-9 y 11meses de edad se reportó una menor distancia recorrida, así como un mayor tiempo de permanencia en el centro del campo abierto84,85. En otro estudio en el mismo modelo transgénico para la EA se ha observado un deterioro en la adquisición de ratones hembra de 8-9meses de edad84, así como un deterioro en la formación de la memoria a largo plazo en ratones hembra y macho de 10meses de edad entrenados en el laberinto acuático de Morris69,72. También se ha visto afectada la memoria en el condicionamiento del miedo y en el laberinto enT86. Con respecto a la conducta innata, en hembras 3xTg-AD de 11meses de edad se ha observado un deterioro en la conducta de construcción de nido87.
Vista holísticaEn los modelos transgénicos, la EA es considerada una patología multifactorial en donde participan muchos elementos en las diferentes etapas de la enfermedad. Además se ha reportado una amplia interacción entre estos factores, lo cual permite que el deterioro cognitivo sea más severo.
El deterioro cognitivo es uno de los síntomas de la EA; sin embargo, esta afectación cognoscitiva resulta de la interacción de múltiples factores, como se menciona en este trabajo, es decir, los efectos del estrés oxidativo y de la respuesta inmune, que inciden directamente en la plasticidad sináptica y estructural, específicamente en los estudios realizados en diferentes modelos transgénicos para la EA.
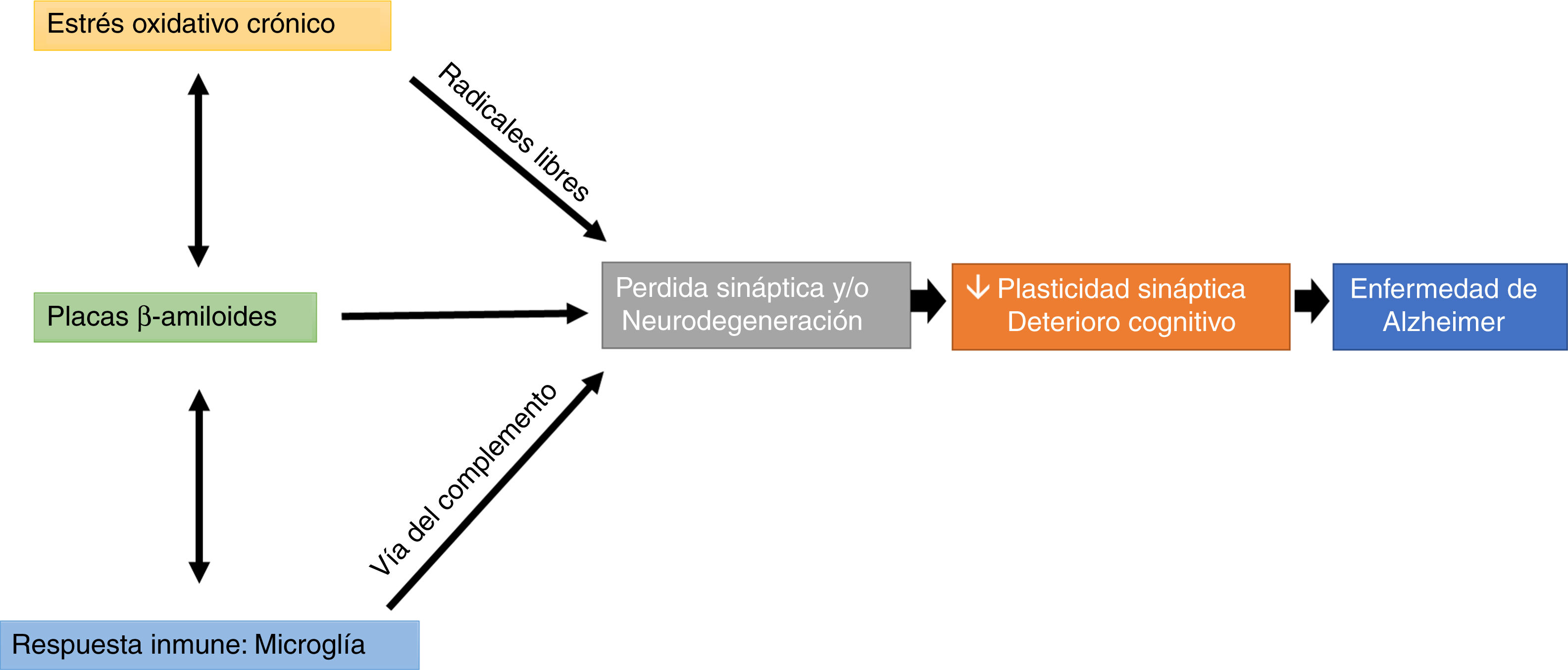
Un factor que llama mucho la atención es la participación de la microglía a través de la vía del complemento en la trogocitosis de las sinapsis, lo cual podría suponer que hay una pérdida de las espinas dendríticas, estos microcompartimentos se han considerado como uno de los elementos clave en la formación de la memoria (fig. 1).
, en la respuesta inmune (trogocitosis por parte de la microglía) y en la formación de placas beta amiloide sobre la pérdida de la plasticidad sináptica y estructural, lo que produce un deterioro cognitivo característico de la enfermedad de Alzheimer.")
Efectos de una alteración en el balance redox (estrés oxidativo crónico), en la respuesta inmune (trogocitosis por parte de la microglía) y en la formación de placas beta amiloide sobre la pérdida de la plasticidad sináptica y estructural, lo que produce un deterioro cognitivo característico de la enfermedad de Alzheimer.
El estrés oxidativo y el sistema inmune pueden modular la aparición y el desarrollo de placas βA, y estas placas pueden afectar directamente la transmisión sináptica, resultando en la afectación de procesos cognitivos (fig. 1).
En el estudio con modelos transgénicos para la EA se han evaluado una amplia gama de paradigmas conductuales en donde se reporta comúnmente el deterioro cognitivo a diferentes edades, es decir, en diferentes etapas de la EA; no obstante, hay ciertos elementos que no se están teniendo en cuenta a la hora de discutir los efectos de esta patología sobre la ejecución de tareas comportamentales, y dentro de estos elementos el más sobresaliente es el estrés. Existen tareas conductuales que inducen una alta liberación de hormonas del estrés, las cuales pueden afectar el aprendizaje y la consolidación de la memoria. Otro de los elementos es la actividad motora: se ha reportado una variedad de trabajos que muestran que la EA es capaz de disminuir el desempeño motor de animales transgénicos para esta enfermedad neurodegenerativa, debido a que existen tareas que requieren un buen desempeño motor; los resultados obtenidos reflejan un deterioro cognitivo, el cual podría estar enmascarado por las deficiencias motoras ya reportadas.
FinanciaciónEl presente trabajo fue financiado por los proyectos de Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT-IN-203616) y al Consejo Nacional de Ciencia y Tecnología (CONACyT CB-255399), Beca de Doctorado a I.V-R (Consejo Nacional de Ciencia y Tecnología 303606).
Conflicto de interesesLos autores declaran no tener conflicto de intereses.
Agradecemos a la técnico de laboratorio Azucena Aguilar Vázquez. Este trabajo fue realizado durante la estancia posdoctoral de P.C. Bello-Medina en el laboratorio de Neuromorfometría y Desarrollo, en el Instituto de Neurobiología de la UNAM.
La trogocitosis ha sido definida en el sistema inmune como un mecanismo no apoptótico para la captura rápida de los componentes de la membrana celular. Difiere de la fagocitosis, ya que esta involucra el engullimiento y la eliminación de estructuras celulares con un tamaño mayor a 1μm48.

