



Worldwide, approximately 50 million people have dementia, with Alzheimer disease (AD) being the most common type, accounting for 60%-70% of cases. Given its high incidence, it is imperative to design studies to expand our knowledge about its onset and development, and to develop early diagnosis strategies and/or possible treatments. One methodological strategy is the use of transgenic mouse models for the study of the factors involved in AD aetiology, which include oxidative stress and the immune response.
DevelopmentWe searched the PubMed, Scopus, and Google Scholar databases for original articles and reviews published between 2013 and 2019. In this review, we address 2 factors that have been studied independently, oxidative stress and the immune response, in transgenic models of AD, and discuss the relationship between these factors and their impact on the loss of synaptic and structural plasticity, resulting in cognitive impairment.
ConclusionThis review describes possible mechanisms by which oxidative stress and the immune response participate in the molecular, cellular, and behavioural effects of AD, observing a close relationship between these factors, which lead to cognitive impairment.
En el mundo, alrededor de 50 millones de personas padecen demencia; la forma más común es la enfermedad de Alzheimer (EA), que representa el 60–70% de los casos. Dada su alta incidencia, se hace imperativo diseñar estudios que permitan ampliar el conocimiento sobre su aparición y desarrollo, para proponer diagnósticos tempranos y/o posibles tratamientos. Una de las estrategias metodológicas que se han desarrollado son los modelos transgénicos murinos para el estudio de los factores involucrados en su etiología, y entre ellos, el estrés oxidativo y la respuesta inmune.
DesarrolloSe realizó una búsqueda de artículos originales y revisiones en PubMed, Scopus y Google Scholar (2013–2019). En esta revisión abordamos dos factores que han sido estudiados de forma independiente: el estrés oxidativo y la respuesta inmune en modelos transgénicos para la EA, y se discute la relación que existe entre ellos y que impacta en la pérdida de la plasticidad sináptica y estructural, produciendo como efecto final el deterioro cognitivo.
ConclusiónEsta revisión describe posibles mecanismos en donde participan el estrés oxidativo y la respuesta inmune sobre los efectos moleculares, celulares y conductuales en la EA, observando una estrecha relación entre estos elementos que conducen hacia el deterioro cognitivo.
Dementia is a syndrome, normally chronic or progressive, that causes cognitive impairment. Nearly 50 million people worldwide have dementia, and the incidence increases every year. Several forms of dementia have been described, with the most common being Alzheimer disease (AD), which accounts for 60%-70% of cases.1
AD is a neurodegenerative disorder characterised by presence in the brain of diffuse neuritic plaques containing beta amyloid protein (Aβ) and neurofibrillary tangles containing hyperphosphorylated tau protein.2
AD is a multifactorial neurodegenerative disease; research is constantly identifying new factors involved in the appearance and progression of the disease. This complicates the study and design of treatments that focus on the symptomatic phase of AD.
The use of transgenic mouse models of AD has provided much information on the appearance, progression, and early and late diagnosis of the disease, as well as on possible prevention strategies, including symptomatic treatments or even the design of new treatments aimed at eliminating this type of dementia. However, the animal models currently available do not replicate all the symptoms observed in humans. In this review of studies with transgenic mouse models, we use the term AD to refer to Alzheimer-type disease in a broader sense.
Over 160 transgenic animal models of AD are currently available, and differ according to the mutated gene and the number of overexpressed proteins. The most frequently used models of AD are shown in Table 1; some are double- or triple-transgenic models based on the metabolism of the amyloid precursor protein (APP), presenilin, or both. Other models are based on tau protein metabolism, and some are triple-transgenic models expressing presenilin/APP/tau (Manzano et al., 2009). Some lines present alterations in apolipoprotein E (ApoE), and are used to study the effects of Aβ oligomers (soluble toxic species).
Articles including transgenic mouse models of AD (3xTg-AD, 5xFAD, Tg2576, APP/PS1, and rTg4510) published on PubMed in the last 5 years (2015-2019). Four of these models are based on the metabolism of the amyloid precursor protein (APP), 3 feature mutations in the PSEN1 gene, and only 2 present a mutation in the MAPT gene.
| Model | Mutated genes or overexpressed proteins | No. articles published | ||||
|---|---|---|---|---|---|---|
| 2019 | 2018 | 2017 | 2016 | 2015 | ||
| 3xTg-AD | APP, PSEN1, MAPT | 10 | 47 | 44 | 39 | 49 |
| 5xFAD | APP, PSEN1 | 12 | 67 | 62 | 50 | 56 |
| Tg2576 score | APP | 8 | 20 | 41 | 42 | 58 |
| APP/PS1 | APP, PSEN1 | 2 | 26 | 19 | 29 | 16 |
| rTg4510 | MAPT | 3 | 12 | 8 | 9 | 6 |
In this review, we focus on the role of oxidative stress and the immune response on AD in different transgenic animal models of the disease. We also analyse the impact of the disease on synaptic plasticity and cognition.
Oxidative stressOxidative stress has been widely linked to neurodegenerative diseases,3–5 including AD.6–9 Oxidative stress and Aβ oligomerisation precede the onset of clinical dementia in AD.10 Studies with several mouse strains serving as transgenic models of AD have revealed increased levels of oxidative stress markers.11
The brain is particularly vulnerable to oxidative stress due to its high demand for oxygen, high levels of polyunsaturated fatty acids, low levels of antioxidants, and relatively high concentration of transition metal ions.12,13
Transgenic mice expressing different mutations linked to AD have enabled research into the role of oxidative stress in the disease. It should be noted that oxidative stress markers (most of which are lipid peroxidation products) are detectable before the appearance of Aβ plaques and cognitive alterations, which suggests that these markers may help in early diagnosis of the condition.7,14
One of the main hallmarks of oxidative stress is lipid peroxidation, which results in the formation of F2-isoprostanes, F4-neuroprostanes, malondialdehyde, and 4-hydroxy-2-nonenal (HNE); these molecules are used as in vivo biomarkers of oxidative stress.14–17 Oxidative stress also causes protein oxidation, the main marker of which is protein carbonylation,18,19 and oxidation of nitrogenous bases, with 8-oxo-2′-deoxyguanosine (8-oxo-dG) and 8-hydroxyguanosine (8-OHG) being the main markers of oxidative stress to DNA and RNA, respectively.20–22
For example, the Swedish APP mutation found in Tg2576 transgenic mice induces an increase in F2-isoprostane levels in the urine, plasma, and brain tissue at 8 months of age, i.e., 2-4 months before the appearance of Aβ plaques and spatial memory impairment. This increase becomes more marked at the age of 12 and 18 months, which has been correlated with onset and progression of Aβ accumulation.23 Furthermore, increased detection of HNE immunoreactivity has been observed in dystrophic neurites adjacent to Aβ plaques.24 Regarding direct oxidation of proteins, increased levels of protein carbonyls have been observed in the hippocampus of transgenic mice.25–27
The Thy1−APP751SL mouse strain presents reduced activity of the Cu/Zn-SOD antioxidant enzyme and increased lipid peroxidation, as detected with the HNE marker. This mouse strain is characterised by overexpression of the 751 isoform of human APP (APP751) and carries the London and Swedish mutations, which induce high expression of APP, high Aβ levels, and onset of plaque formation at the age of 6 months.28
The PDGF-APP695SDL line, which presents the 695 isoform and carries the Swedish, Dutch, and London mutations, presents lower Aβ plaque load, with Aβ accumulation being observable until the age of 18 months.28 This mouse strain does not present decreased Cu/Zn-SOD activity or increased lipid peroxidation, which suggests that the decrease in antioxidant activity that leads to oxidative stress is linked to β-amyloidogenic cleavage of APP.29
In transgenic mice carrying PSEN1, the gene has been shown to induce early Aβ42 accumulation in senile plaques.30 Mice expressing human mutant PS1 present increased levels of oxidative stress markers, protein carbonylation, and HNE.31 Furthermore, APP/PSEN1 double-transgenic mice present higher levels of these markers, as well as increased F2-isoprostane levels.31,32
PS19 transgenic mice have been used extensively in the study of tauopathy in AD, and express the P301S mutant form of the human tau protein. They present iron overload in the cortex and hippocampus at 7.5 months of age.33 Iron accumulation has been linked to hyperphosphorylated tau aggregation and redox imbalance.34 Furthermore, PS19 transgenic mice also present decreased expression of antioxidant enzymes GPx4, xCT, and SOD1, and decreased SOD activity.33 Research has also shown that transgenic mice expressing human tau protein present increased protein carbonylation in the cerebral cortex and increased levels of 8-oxo-dG, a marker of oxidative damage to nucleic acids.35
Triple-transgenic mice (3xTg-AD) expressing PS1(M146V), APP(Swe), and tau(P301L) transgenes36 have been shown to present lower levels of glutathione and vitamin E and increased lipid peroxidation. They also present greater glutathione peroxidase and superoxide dismutase activity. These alterations are detectable during Aβ oligomerisation and before the appearance of Aβ plaques and neurofibrillary tangles, indicating that oxidative stress occurs early in the course of the disease.37
A recent study also observed that the hippocampus of 3xTg-AD mice presents a twofold increase in levels of reactive oxygen species and reactive nitrogen species, which explains the accumulation of such markers of lipid peroxidation as HNE, malondialdehyde, and 8-iso-PGF2.38 In terms of protein damage, increased protein carbonylation and tyrosine nitration have been reported.19,38 Lastly, research has also shown an increase in the levels of 8-oxo-dG and 8-OHG (markers of DNA and RNA oxidative damage, respectively).38
Immune response and microgliaThe role of inflammation in AD was first described, almost simultaneously, by Oskar Fischer and Alois Alzheimer, although for many years little attention was paid to the study of the immune response in the central nervous system of these patients. In recent years, this topic has become increasingly important,39 as new evidence suggests that inflammation plays a causal role in AD, with its contribution to disease pathogenesis being equally or even more significant than that of tau and Aβ.40
Genome-wide association studies have found an association between the microglial immune response and AD.41 Most of the genes identified as risk factors are present in the microglia, as is the case with TREM2 and APOE.41,42
The microglia may present a proinflammatory response in AD: post mortem studies have shown that the morphology of microglial cells associated with Aβ plaques and/or neurofibrillary tangles is similar to that of microglial cells in such proinflammatory events as sepsis. Cells presenting morphological alterations are known as dystrophic or senescent microglia.43 It has been suggested that dystrophic microglia, rather than proinflammatory microglia, may be related to synaptic dysfunction and neurodegeneration in AD.43 The microglial phenotype of the 5xFAD transgenic mouse model differs from the proinflammatory phenotype, and has been termed DAM (disease-associated microglia); this phenotype is characterised by TREM2 and APOE overexpression.44 Likewise, APP/PS1 transgenic mice are reported to present a different microglial phenotype from the proinflammatory phenotype near Aβ plaques; this phenotype has been termed MGnD (microglial neurodegenerative phenotype). These microglial phenotypes observed in the 3 transgenic mouse models of AD overexpress TREM2 and APOE,45 which have been associated with increased risk of developing AD.46 It should be noted that the expression of these genes is inhibited by the proinflammatory microglial phenotype induced by lipopolysaccharide or interferon-γ.45
During postnatal development, microglia play a significant role in eliminating neurons that do not belong to functional circuits and in remodelling synapses via the C1q and C3 proteins of the complement pathway.47 Unused synapses are tagged by these complement pathway proteins and subsequently recognised by microglial complement receptor 3 (CR3) for clearance.47 This process is known as trogocytosis,† and only affects presynaptic boutons and axons.48 This finding is interesting since the main characteristic of AD is presence of Aβ plaques and neurofibrillary tangles; other characteristics include synaptic loss, cognitive impairment, and neurodegeneration in advanced stages,49 in which microglia are also thought to play a role.
Amyloidogenic models such as 5xFAD transgenic mice display synaptic dysfunction, synaptic loss, and neuronal death.50 However, microglial elimination prevents the loss of dendritic spines and neurons without changes in Aβ levels or Aβ plaque size, which suggests that the microglial phenotype observed in this animal model is responsible for dendritic spine loss and neuronal death.42,51 Aβ has been found to play a major role in dendritic pruning, since DAM and MGnD phenotypes are observed in advanced stages of AD in amyloidogenic mouse models, but not in non-transgenic mice.44,45 Furthermore, Aβ oligomers can activate the complement pathway, causing C1q to tag synapses for subsequent recognition by CR3 and elimination by microglia.52
Aβ plaques have been shown to contain complement pathway proteins39; in non-transgenic mice, C3 and CR3 favour Aβ phagocytosis by microglial cells.53 This suggests that complement pathway proteins tag Aβ plaques, making them recognisable by microglia for subsequent clearance.
Such amyloidogenic models as APP transgenic mice have shown that CR3 decreases microglial phagocytic activity54; ablation of C3 in APP/PSI mice prevents synaptic loss, despite increased Aβ plaque load.55 The differences observed between non-transgenic models and APP/PS1 transgenic models may be explained by the microglial phenotype; this suggests that C3 and CR3 may protect neurons in the proinflammatory phenotype but promote synaptic loss and neurodegeneration in the DAM and MGnD phenotypes.
Interestingly, the DAM and MGnD phenotypes overexpress TREM2 in the vicinity of Aβ plaques,44,45 since this receptor displays a preference for Aβ oligomers,46 which may induce synaptic elimination via the complement pathway.52
One important question remains: why do microglia lose homeostasis and eliminate synapses in an uncontrolled manner? A plausible hypothesis is that the gene encoding C-X3-C motif chemokine receptor 1 (CX3CR1) is involved in this process; this gene inhibits microglia and its expression is reduced in early stages in amyloidogenic transgenic models, which may cause TREM2 overexpression in advanced stages of the disease.44
Microglia have also been found to play a major role in the MAPT P301S transgenic model (whose main characteristic is the development of tauopathy), since clearance of this cell type and of senescent astrocytes prevents fibrillary tangle deposition and neurodegeneration.56 This suggests that, as occurs in amyloidogenic models, some microglial phenotype other than the proinflammatory phenotype is involved in AD.
Synaptic plasticity and cognitionSynaptic plasticity is the mechanism that induces changes in synaptic efficiency as a result of high-frequency, experience-dependent neuronal activity.57 It involves a wide range of structural and functional changes, including long-term potentiation, synaptogenesis, axonal and dendritic remodelling, and in some cases adult neurogenesis.58
Some of these mechanisms of structural synaptic plasticity involve synaptic remodelling, that is, structural changes in dendrites, specifically in dendritic spines. Dendritic spines are cellular compartments mainly composed of cytoskeletal elements.59 Their internal structure contains a protein complex termed postsynaptic density, which anchors a wide range of molecules, including such neurotransmitter receptors as the AMPA, NMDA, and kainate receptors.60 The morphology of dendritic spines varies, and is classified into different types, including thin, mushroom, and stubby spines.61 Dendritic spine morphology is linked to their function: thin spines have been associated with such cognitive processes as learning, whereas mushroom spines have been associated with memory62 and stubby spines are thought to regulate excitability by regulating calcium ions.63
Such brain structures as the hippocampus, amygdala, striatum, and cortex participate actively in information processing during learning and memory processes; these cognitive processes induce changes in synaptic plasticity in normal conditions,64–68 but are impaired in patients with AD and transgenic models of the disease. Transgenic models have been used to study the impact of AD on synaptic plasticity at the molecular, cellular, and cognitive/behavioural levels.
At the molecular level, 3-month-old female 3xTg-AD mice have been observed to display increased expression of GluA3 and GluA4 in the hippocampus. However, expression decreased at the age of 12 months, as compared to non-transgenic mice,69 which suggests that the glutamatergic transmission system is impaired in AD. Likewise, altered expression of the activity-regulated cytoskeleton-associated protein (Arc) has been observed in the dorsal hippocampus of 3xTg-AD mice. Arc has been used as a marker of neuronal activity,70 since the gene encoding the protein is an immediate-early gene, as well as a marker of synaptic plasticity due to its involvement in AMPA receptor endocytosis and synaptic scaling.71 Arc expression is increased in the hippocampal CA3 region of 10-month-old male 3xTg-AD mice. However, when mice are trained in the Morris water maze, Arc expression does not change with respect to baseline values; furthermore, most Arc-expressing cells are neurons presenting Aβ plaques.72 Recent evidence suggests that Arc plays a relevant role in synaptopathy in AD.73
A study with one-month-old male 3xTg-AD mice showed that tau phosphorylation is associated with changes in hippocampal theta oscillations and decreases the excitability of neurons in the CA1 region and subiculum.74 Other studies with this animal model have also shown long term potentiation deficits secondary to the formation of Aβ plaques and fibrillary tangles.75 A study including 3xTg-AD mice aged 12-16 months found an increase in voltage-dependent Ca2+ currents in the hippocampal CA1 region, which increased Ca2+ concentration within neurons,76 resulting in neuronal excitotoxicity.77,78
Neurotransmission activates multiple specific signalling cascades, depending on the receptor activated and the experience of the individual. Some of these signalling pathways induce the expression of genes that encode structural proteins, which are responsible for dynamic restructuring of neurons, as is the case with dendritic spines.
In 13-month-old AβPP/PS1 transgenic mice, AD has been observed to alter the levels of such proteins as ERK44, pERK44, c-Fos, mTOR, and AβPP in the parietal cortex and the CA1 region and dentate gyrus of the dorsal hippocampus.79 This indicates impairment of the signalling cascades involving these proteins; as a result, downstream molecular signalling is also altered. This has an indirect impact on molecules involved in structural synaptic plasticity, such as Ca2+/calmodulin/CaMKII/CaV1.2,80 which affect such cytoskeletal proteins as F-actin. This is another pathway that may interfere in structural synaptic plasticity; as previously mentioned, microglial trogocytosis also causes synaptic loss, which may lead to dendritic spine loss.
In APP/PS1 transgenic mice of 1-2 months of age, Aβ protein has been found to induce F-actin depolymerisation, which in turn decreases dendritic spine density.81 Likewise, 4-month-old APP23 and APPswe/PS1dE9 transgenic mice show lower dendritic spine density near Aβ plaques and a decrease in the number of mushroom spines, which are linked to such cognitive processes as memory.82
In animal models of AD, synaptic dysfunction has been linked to cognitive impairment. 3xTg-AD mice have been observed to perform poorly in innate and learned behavioural paradigms. Regarding learned behaviour, these animals spend less time in the open arms of the elevated plus maze and present a lower object discrimination index, as well as long-term retention deficits in the Morris water maze.83 Studies with female 3xTg-AD mice aged 8-9 and 11 months old have reported shorter distance travelled in the open field and longer time spent in inner square.84,85 In this transgenic model, researchers also report poorer acquisition in female mice of 8-9 months of age,84 and impaired long-term memory formation in 10-month-old male and female mice trained in the Morris water maze.69,72 Memory has also been found to be impaired in fear conditioning and T-maze paradigms.86 Regarding innate behaviour, 11-month-old female 3xTg-AD mice have been observed to display alterations in nesting behaviour.87
ConclusionsIn transgenic animal models, AD is considered a multifactorial disease involving numerous elements at different stages. Significant interaction is also reported between these factors, aggravating cognitive impairment.
Cognitive impairment is a typical symptom of AD that results from the interaction between oxidative stress and the immune response, which have a direct impact on structural synaptic plasticity, specifically in studies with different transgenic models of AD.
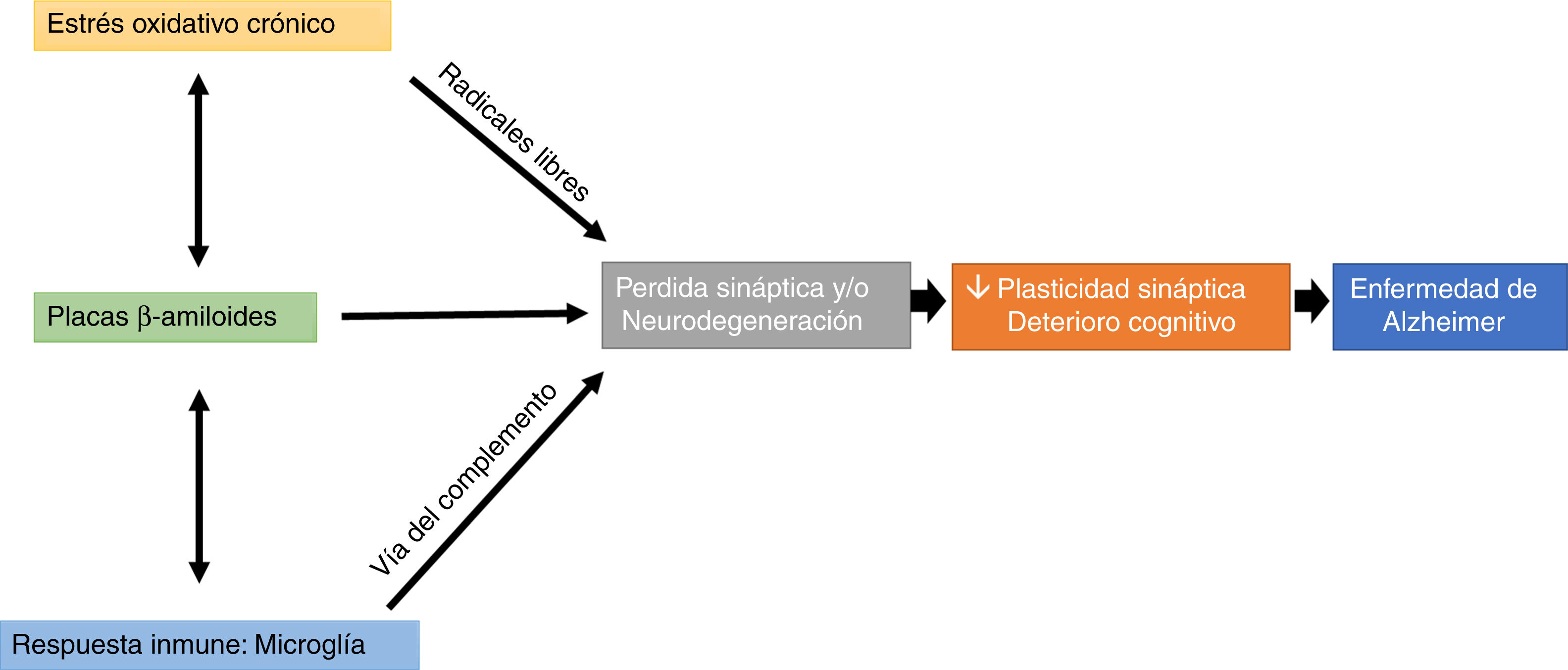
Interestingly, microglial cells are involved in this process, through complement-mediated synaptic trogocytosis; this may entail a loss of dendritic spines, a key element in memory formation (Fig. 1).
, immune response (microglial trogocytosis), and Aβ plaque formation on loss of structural synaptic plasticity, which results in a pattern of cognitive impairment characteristic of Alzheimer disease.")
Oxidative stress and the immune system may modulate the appearance and development of Aβ plaques, which directly affect synaptic transmission, altering cognitive processes (Fig. 1).
Studies with transgenic models of AD have evaluated multiple behavioural paradigms, frequently reporting cognitive impairment at different ages (i.e., at different stages of the disease). However, some elements have not been considered in the assessment of the effect of AD on behavioural task performance, with the most relevant being stress. Some behavioural tasks induce the release of high levels of stress hormones, which may interfere with learning and memory consolidation. Another element is motor activity: multiple studies have shown that AD has a negative impact on motor function in transgenic animal models. The results of these studies reflect cognitive impairment, which may be masked by the motor deficits reported.
FundingThis study was funded by the Support Programme for Research and Technological Innovation (PAPIIT-IN-203616) and by the Mexican National Council of Science and Technology (CONACyT CB-255399), through a doctoral scholarship to IVR (CONACyT 303606).
Conflicts of interestThe authors have no conflicts of interest to declare.
We wish to thank laboratory technician Azucena Aguilar Vázquez. This study was conducted during PCBM’s post-doctorate fellowship at the Laboratory of Neuromorphometry and Development of the UNAM’s Neurobiology Institute.
Trogocytosis has been described in the immune system as a non-apoptotic mechanism for the rapid capture of cell membrane components. It differs from phagocytosis in that the former involves engulfment and elimination of cellular structures measuring >1 µm.48
Please cite this article as: Bello-Medina PC, González-Franco DA, Vargas-Rodríguez I, Díaz-Cintra S. Estrés oxidativo, respuesta inmune, plasticidad sináptica y cognición en modelos transgénicos de la enfermedad de Alzheimer. Neurología. 2022;37:682–690.
recomendados

Neurología (English Edition) sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas