



Las mucopolisacaridosis (MPS) son un grupo de enfermedades hereditarias de depósito lisosomal. El objetivo de esta revisión es describir las alteraciones neurorradiológicas en los niños evaluados en nuestro hospital con este diagnóstico, buscar la posible correlación de estas alteraciones con el tipo de MPS y con la gravedad clínica, y comparar nuestros hallazgos con lo descrito en la literatura.
Material y métodosRevisamos retrospectivamente las historias clínicas de 19 pacientes diagnosticados de MPS en el periodo 1992-2010: 7 tipo I (5 con síndrome de Hurler y 2 con Hurler-Scheie), 10 tipo II o síndrome de Hunter (4 con la forma grave y 6 con la moderada), 1 tipo III o síndrome de Sanfilippo y 1 tipo VI o síndrome de Maroteaux-Lamy. Se analizaron las pruebas de neuroimagen: tomografía computarizada (TC) en 5 pacientes y resonancia magnética craneal (RMC) en 15.
ResultadosEncontramos un amplio espectro de alteraciones radiológicas. En la TC destaca la megacisterna magna (3/5, 60%); en la RMC el aumento de los espacios perivasculares (11/15, 73%), la alteración parcheada de la sustancia blanca (SB) (11/15, 73%) y la ventriculomegalia (5/15, 33%).
ConclusionesAlgunas anomalías neurorradiológicas son frecuentes en las MPS (aumento de los espacios perivasculares, alteraciones de la SB, ventriculomegalia), por lo que ante estos hallazgos debemos investigar esta posibilidad diagnóstica, especialmente en pacientes con clínica compatible. No hemos hallado datos específicos de cada tipo de MPS, ni relación de estas alteraciones radiológicas con la gravedad de la forma clínica.
Mucopolysaccharidoses (MPS) are a group of inherited disorders due to lysosomal enzyme deficiencies. The aims of this study are to describe the neuroimaging findings in children evaluated in our hospital with this diagnosis, looking for a possible correlation of these alterations with the type of MPS and clinical severity, and finally to compare these findings with those previously reported.
Material and methodsWe retrospectively analysed the medical records of 19 patients who had been diagnosed with MPS between 1992 and 2010: 7 had type I (5 with Hurler syndrome and 2 with Hurler-Scheie syndrome), 10 had type II or Hunter syndrome (4 with the severe form and 6 with the mild form), 1 had type III or Sanfilippo syndrome and 1 had type VI or Maroteaux-Lamy syndrome. We assessed the brain neuroimaging studies: computed axial tomography (CAT) in 5 patients, and magnetic resonance imaging (MRI) in 15.
ResultsWe observed a broad spectrum of neuroimaging anomalies. In CAT: mega cisterna magna (3/5, 60%). In brain MRI: dilated Virchow-Robin perivascular spaces (11/15, 73%), white matter abnormalities (11/15, 73%), and ventriculomegaly (5/15, 33%).
ConclusionsAbnormal findings in neuroimaging studies are frequent in MPS (dilated Virchow-Robin perivascular spaces, white matter abnormalities and ventriculomegaly). Thus, given these abnormalities we should be aware of this possible diagnosis, particularly when typical signs and symptoms are present. However, we did not find a correlation between these findings and either any specific type of MPS or clinical severity.
Las mucopolisacaridosis (MPS) son un grupo de errores congénitos del metabolismo caracterizados por un déficit en una de las enzimas lisosomales responsables de la degradación de los glucosaminoglucanos (GAG) o mucopolisacáridos. Este déficit conlleva la acumulación anormal de GAG dentro de los lisosomas, así como su excreción elevada en la orina. Existen 7 tipos diferentes de MPS y la prevalencia de dichas enfermedades en conjunto se estima en 1/22.500 individuos. Son enfermedades multisistémicas degenerativas, crónicas y progresivas, que incluyen numerosas alteraciones, tanto físicas como mentales1,2. Generalmente son difíciles de detectar en el recién nacido (salvo que haya otros familiares afectados), puesto que estos niños suelen tener una apariencia normal al nacimiento. Progresivamente desarrollan diversas alteraciones fenotípicas entre las que destacan las siguientes: cara tosca característica, alteraciones en el sistema esquelético (disostosis múltiple), talla baja, contracturas, afectación cardiovascular (valvulopatía, miocardiopatía), visceromegalias, hernias inguinales y umbilicales, afectación de la piel por infiltración, opacidad corneal y sordera2-4. En cuanto a las manifestaciones neurológicas, estas son muy diferentes según el tipo de MPS. En las formas graves de los tipos I, II y VII, y en el tipo III es frecuente el deterioro cognitivo progresivo5-7. Los pacientes con neurodegeneración presentan gran discapacidad y mueren en la primera o segunda década de la vida. Aquellos pacientes con formas más atenuadas pueden llegar a la adolescencia o a la vida adulta, aunque frecuentemente con gran morbilidad5. En lo que a la herencia se refiere, todos los tipos se transmiten de forma autosómica recesiva, salvo la MPS tipo II o síndrome de Hunter, que se hereda de forma recesiva ligada a X. El diagnóstico de las MPS se basa en el análisis cuantitativo de los GAG en la orina del paciente. Actualmente, además, se procede al estudio enzimático específico en cada forma de MPS y al estudio molecular. El tratamiento de las MPS es complejo5. El trasplante de progenitores hematopoyéticos se ha mostrado eficaz en algunas MPS y en la actualidad es el tratamiento de elección en algunos pacientes con la forma grave de MPS I, pero presenta una alta morbilidad y mortalidad2. La terapia de sustitución enzimática ha mostrado ser eficaz en las MPS I, II y VI. En todas las MPS se deberán atender adecuadamente las complicaciones que surjan con el avance de la enfermedad2,5,6.
Los primeros estudios neurorradiológicos en la evaluación de las MPS se realizaron con tomografía computarizada (TC), observándose datos no específicos de estas enfermedades. Ejemplos de estos hallazgos en la TC son: áreas hipodensas en la sustancia blanca (SB) y dilatación ventricular y de los espacios subaracnoideos. Estudios posteriores demostraron que la resonancia magnética cerebral (RMC) es más sensible que la TC y ofrece información más detallada de las alteraciones del sistema nervioso central8.
El objetivo de esta revisión es describir las alteraciones neurorradiológicas en los niños evaluados en nuestro hospital con diagnóstico de alguno de los tipos de MPS, buscar la posible correlación de estas alteraciones con el tipo de MPS y con la gravedad de la forma clínica, y comparar nuestros resultados con lo descrito en la literatura.
Pacientes y métodosSe revisaron retrospectivamente las historias clínicas de 19 pacientes diagnosticados de MPS o evaluados por este motivo en nuestro servicio entre los años 1992 y 2010. El diagnóstico se realizó inicialmente por la excreción urinaria aumentada de GAG y con la medición de la actividad de la enzima deficitaria correspondiente y, siempre que fue posible, el estudio genético. Según esto, clasificamos a los pacientes en los diferentes tipos de MPS. Además, en función de la edad de presentación de los síntomas y la evolución de la clínica neurológica, estos enfermos se subdividen en formas graves y atenuadas, con un pronóstico más o menos desfavorable. Los 19 pacientes estudiados se subdividieron en: 7 pacientes tipo I (de los cuales 5 presentan formas graves o síndrome de Hurler y 2 formas intermedias o síndrome de Hurler-Scheie), 10 tipo II o síndrome de Hunter (4 con una forma grave y 6 con una forma moderada), 1 tipo III o síndrome de Sanfilippo y 1 tipo VI o síndrome de Maroteaux-Lamy.
Los 4 primeros pacientes de nuestra serie fueron estudiados únicamente con TC craneal como prueba de neuroimagen. Los 4 son MPS tipo II o Hunter (3 graves y 1 moderado). En los 15 pacientes restantes se llevó a cabo RMC: 5 Hurler, 2 Hurler-Scheie, 1 Hunter grave, 5 Hunter moderados, 1 Sanfilippo y 1 Maroteaux-Lamy. A uno de los pacientes con síndrome de Hurler-Scheie, inicialmente también se le realizó una TC craneal como prueba de imagen.
Para la realización de la RMC se emplearon secuencias ponderadas en T1 y T2, en plano sagital y axial, y se valoró principalmente la presencia o ausencia de ventriculomegalia o, en su caso, de hidrocefalia, alteración parcheada de la SB, aumento de los espacios perivasculares de Virchow-Robin (EPVR), lesiones del cuerpo calloso, atrofia cortical o subcortical, así como alteraciones de la fosa posterior. Las imágenes fueron evaluadas por radiólogos expertos, que no contaban con datos referentes a la situación neurológica de los pacientes, tan sólo en algunos casos disponían del tipo de MPS concreto. De los 15 pacientes a los que se realizó RMC, 5 cuentan con dos o más pruebas de imagen durante el seguimiento de la enfermedad. Del resto disponemos de una única prueba realizada generalmente en las primeras semanas tras el diagnóstico o la sospecha clínica.
Una vez diagnosticados, los pacientes seguían valoraciones clínicas en las subespecialidades pediátricas correspondientes semestral o anualmente (neuropediatría, otorrinolaringología, traumatología, cardiología, oftalmología…). En la última década, a aquellos pacientes que habían hecho el seguimiento clínico completo de la enfermedad en nuestro centro también se les aplicó el protocolo de valoración neuropsicológica consensuado para este tipo de enfermedades9.
ResultadosEn todos los pacientes de nuestra serie se halló algún tipo de alteración en la neuroimagen, tanto en TC craneal como en RMC. En los 5 pacientes a los que se realizó una TC destacó como dato más frecuente la megacisterna magna, que se objetivó en 3 de ellos (60%), imágenes hipodensas de la SB (en 2 pacientes), un quiste del septum pellucidum y una agenesia parcial posterior del cuerpo calloso.
En la RMC los hallazgos más frecuentes fueron: el aumento de los EPVR (11/15, 73%), la alteración parcheada de la SB (11/15, 73%) y la ventriculomegalia (5/15, 33%). En 3 de los pacientes se halló atrofia subcortical (en uno de los pacientes con síndrome de Hurler-Scheie, en otro con síndrome de Hurler y en el paciente con síndrome de Sanfilippo). En algunos pacientes se encontraron alteraciones de fosa posterior: discreta reducción del espacio subaracnoideo anterior y posterior en la unión bulbomedular en el paciente con MPS VI, megacisterna magna en dos niños (uno con síndrome de Hunter moderado y otro con Hurler) y un quiste retrocerebeloso en un paciente con Hunter moderado. Se observaron alteraciones del cuerpo calloso en 5 pacientes: lesiones focales en un paciente con síndrome de Hurler, aumento de los EPVR en 2 niños con síndrome de Hunter (uno moderado y otro grave) y atrofia del cuerpo calloso en el paciente afectado de MPS tipo III y en un niño con síndrome de Hunter moderado.
En el seguimiento, a 5 pacientes se les realizó una RM cervical y a 5 una RM de columna completa o lumbosacra. Se encontró algún tipo de alteración a nivel cervical alto en 5: 2 niños con Hurler-Scheie, 1 con Hunter grave, 1 con Hunter moderado y 1 con Maroteaux-Lamy. En una de las niñas afectadas de síndrome de Hurler-Scheie se encontró un aumento de partes blandas retroodontoideas y rectificación de la lordosis cervical fisiológica, con disminución del calibre del canal medular a nivel cervical alto. Y en otro paciente afectado de MPS tipo II moderada se halló un engrosamiento leve de partes blandas por detrás de la apófisis odontoides, pero sin disminución significativa del calibre del conducto raquídeo. Otros hallazgos aislados fueron: deformidad del cuerpo de la tercera vértebra cervical (en un niño con Hunter grave) e hipoplasia de la apófisis odontoides (en el paciente con MPS VI y en una de las niñas con Hurler-Scheie). En los 5 niños a los que se realizó RM de columna lumbosacra se encontró acuñamiento anterior, posterior o deformidad en la primera o segunda vértebra lumbar.
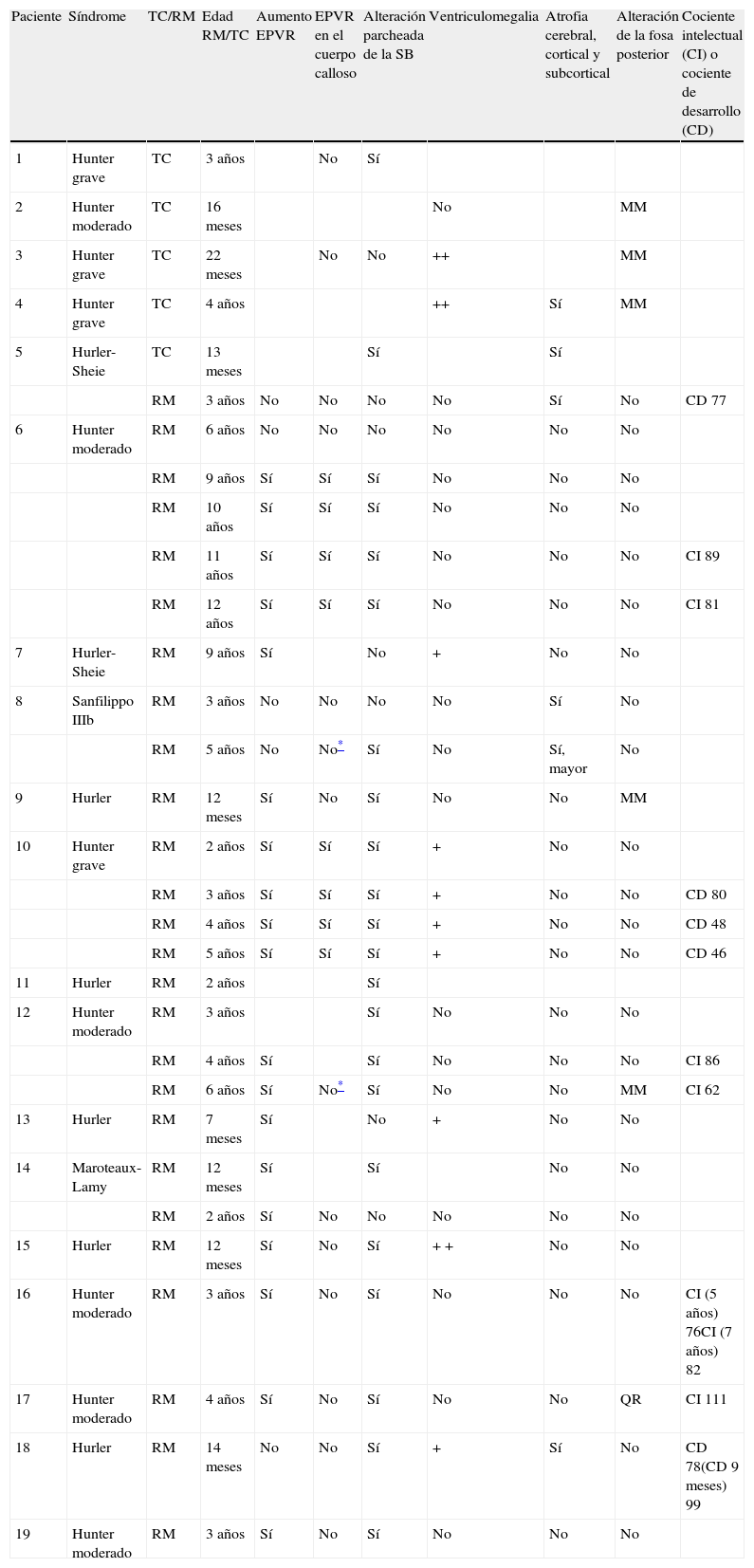
En las MPS I y II, las más frecuentes en nuestra serie, no se halló relación de las alteraciones neurorradiológicas con cada tipo de MPS en concreto, ni tampoco con la gravedad de la forma clínica. En los últimos pacientes estudiados en nuestro centro, con estudio neuropsicológico completo, tampoco se objetivó relación entre las alteraciones radiológicas y su nivel de desarrollo cognitivo, establecido con las escalas Weschler10,11 a partir de los 3 años o con las escalas de desarrollo de Batelle12 sin son menores o están muy deteriorados (tabla 1).
Hallazgos radiológicos de los pacientes incluidos en el estudio
| Paciente | Síndrome | TC/RM | Edad RM/TC | Aumento EPVR | EPVR en el cuerpo calloso | Alteración parcheada de la SB | Ventriculomegalia | Atrofia cerebral, cortical y subcortical | Alteración de la fosa posterior | Cociente intelectual (CI) o cociente de desarrollo (CD) |
| 1 | Hunter grave | TC | 3 años | No | Sí | |||||
| 2 | Hunter moderado | TC | 16 meses | No | MM | |||||
| 3 | Hunter grave | TC | 22 meses | No | No | ++ | MM | |||
| 4 | Hunter grave | TC | 4 años | ++ | Sí | MM | ||||
| 5 | Hurler-Sheie | TC | 13 meses | Sí | Sí | |||||
| RM | 3 años | No | No | No | No | Sí | No | CD 77 | ||
| 6 | Hunter moderado | RM | 6 años | No | No | No | No | No | No | |
| RM | 9 años | Sí | Sí | Sí | No | No | No | |||
| RM | 10 años | Sí | Sí | Sí | No | No | No | |||
| RM | 11 años | Sí | Sí | Sí | No | No | No | CI 89 | ||
| RM | 12 años | Sí | Sí | Sí | No | No | No | CI 81 | ||
| 7 | Hurler-Sheie | RM | 9 años | Sí | No | + | No | No | ||
| 8 | Sanfilippo IIIb | RM | 3 años | No | No | No | No | Sí | No | |
| RM | 5 años | No | No* | Sí | No | Sí, mayor | No | |||
| 9 | Hurler | RM | 12 meses | Sí | No | Sí | No | No | MM | |
| 10 | Hunter grave | RM | 2 años | Sí | Sí | Sí | + | No | No | |
| RM | 3 años | Sí | Sí | Sí | + | No | No | CD 80 | ||
| RM | 4 años | Sí | Sí | Sí | + | No | No | CD 48 | ||
| RM | 5 años | Sí | Sí | Sí | + | No | No | CD 46 | ||
| 11 | Hurler | RM | 2 años | Sí | ||||||
| 12 | Hunter moderado | RM | 3 años | Sí | No | No | No | |||
| RM | 4 años | Sí | Sí | No | No | No | CI 86 | |||
| RM | 6 años | Sí | No* | Sí | No | No | MM | CI 62 | ||
| 13 | Hurler | RM | 7 meses | Sí | No | + | No | No | ||
| 14 | Maroteaux-Lamy | RM | 12 meses | Sí | Sí | No | No | |||
| RM | 2 años | Sí | No | No | No | No | No | |||
| 15 | Hurler | RM | 12 meses | Sí | No | Sí | + + | No | No | |
| 16 | Hunter moderado | RM | 3 años | Sí | No | Sí | No | No | No | CI (5 años) 76CI (7 años) 82 |
| 17 | Hunter moderado | RM | 4 años | Sí | No | Sí | No | No | QR | CI 111 |
| 18 | Hurler | RM | 14 meses | No | No | Sí | + | Sí | No | CD 78(CD 9 meses) 99 |
| 19 | Hunter moderado | RM | 3 años | Sí | No | Sí | No | No | No |
EPVR: espacios perivasculares de Virchow-Robin; MM: megacisterna magna; QR: quiste retrocerebeloso; +: ligera; ++: moderada-grave.
Las MPS son un grupo heterogéneo de enfermedades con afectación somática y, en algunos casos, neurodegeneración. El diagnóstico de sospecha se realiza en base a los signos y síntomas clínicos característicos, la historia familiar y el aumento de excreción de GAG en orina, y se confirma con el estudio enzimático y genético2,5. La RM resulta una técnica útil para valorar las alteraciones a nivel cerebral y espinal: alteraciones de la SB, hidrocefalia y compresión medular3,13,14. Las alteraciones neurorradiológicas a nivel del parénquima cerebral en las MPS han sido descritas previamente, aunque no existe una amplia bibliografía al respecto, puesto que globalmente son enfermedades poco prevalentes y la RMC no ha sido una prueba ampliamente extendida hasta hace unos años8. Por ello es difícil concluir si existe alguna relación entre estas alteraciones y el tipo de MPS o la gravedad de la forma clínica.
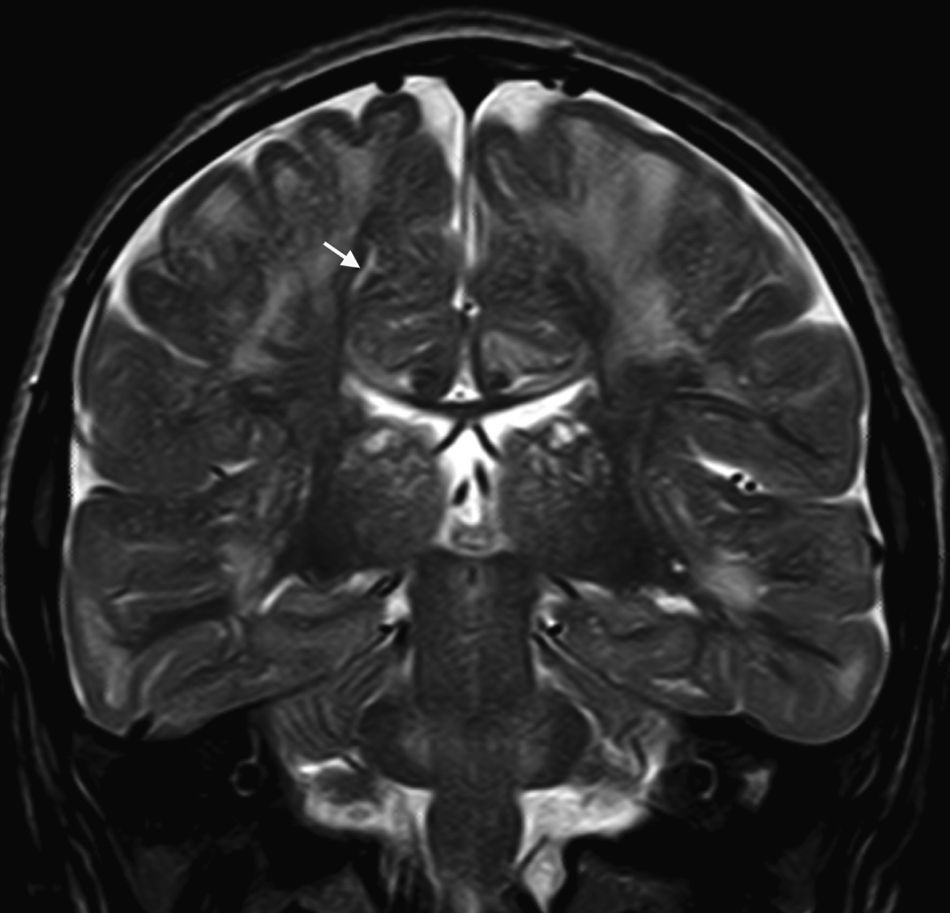
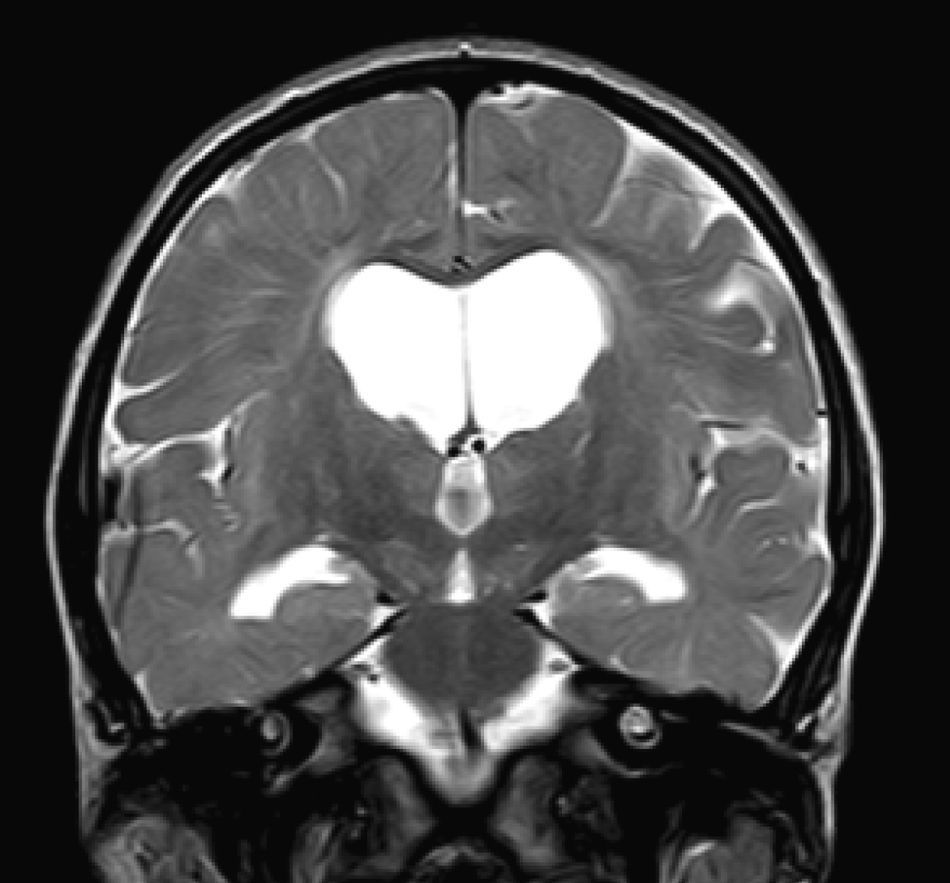
En nuestra serie el hallazgo predominante en la TC es la megacisterna magna (3/5, 60%). En la RMC los hallazgos más frecuentes son: el aumento de los EPVR (11/15, 73%), la afectación parcheada de la SB (11/15, 73%) y la ventriculomegalia (5/15, 33%), lo que coincide con lo publicado en la literatura3,8,13-17). Toshiyuki Seto et al.13 observan que el aumento de los EPVR se encuentra con más frecuencia y severidad en la MPS II, la alteración parcheada de la SB predomina sobre todo en MPS I y II, y la ventriculomegalia se objetiva especialmente en los tipos I y II. Nosotros hemos hallado aumento de EPVR en MPS I, II y VI (fig. 1), al igual que dicho autor, y alteraciones parcheadas de la SB en los tipos I, II y VI, así como ventriculomegalia en las MPS tipo I y II (fig. 2). Por tanto, los hallazgos neurorradiológicos hallados en nuestro grupo de pacientes son bastante característicos, pero ninguno es específico de un tipo concreto de MPS15-17.
. Se identifican cambios de señal hiperintensos en T2 en la sustancia blanca supratentorial y marcada dilatación de espacios perivasculares de Virchow-Robin (flecha) en paciente con síndrome de Hunter.")
. Se identifica ventriculomegalia supratentorial y dilatación de espacios perivasculares de Virchow-Robin en paciente con síndrome de Hunter grave a los 5 años y medio de edad.")
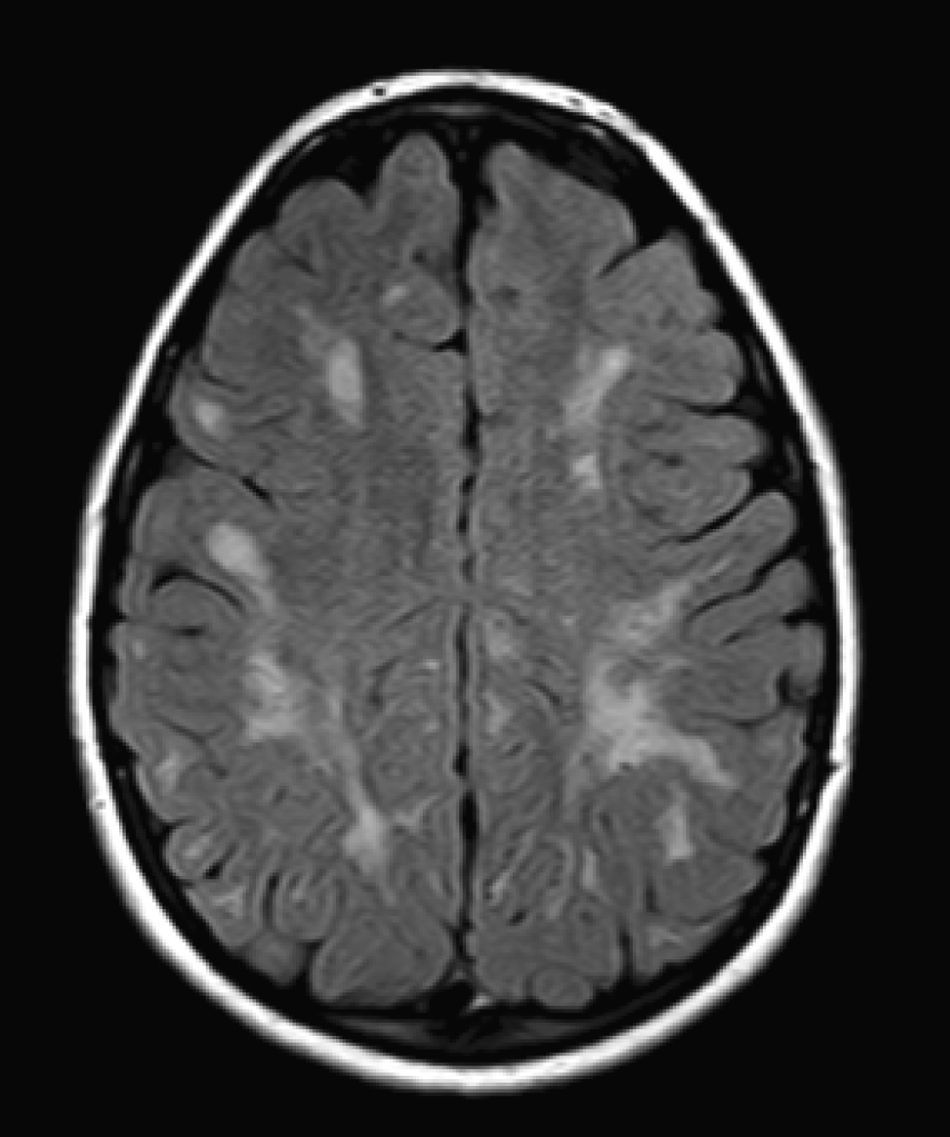
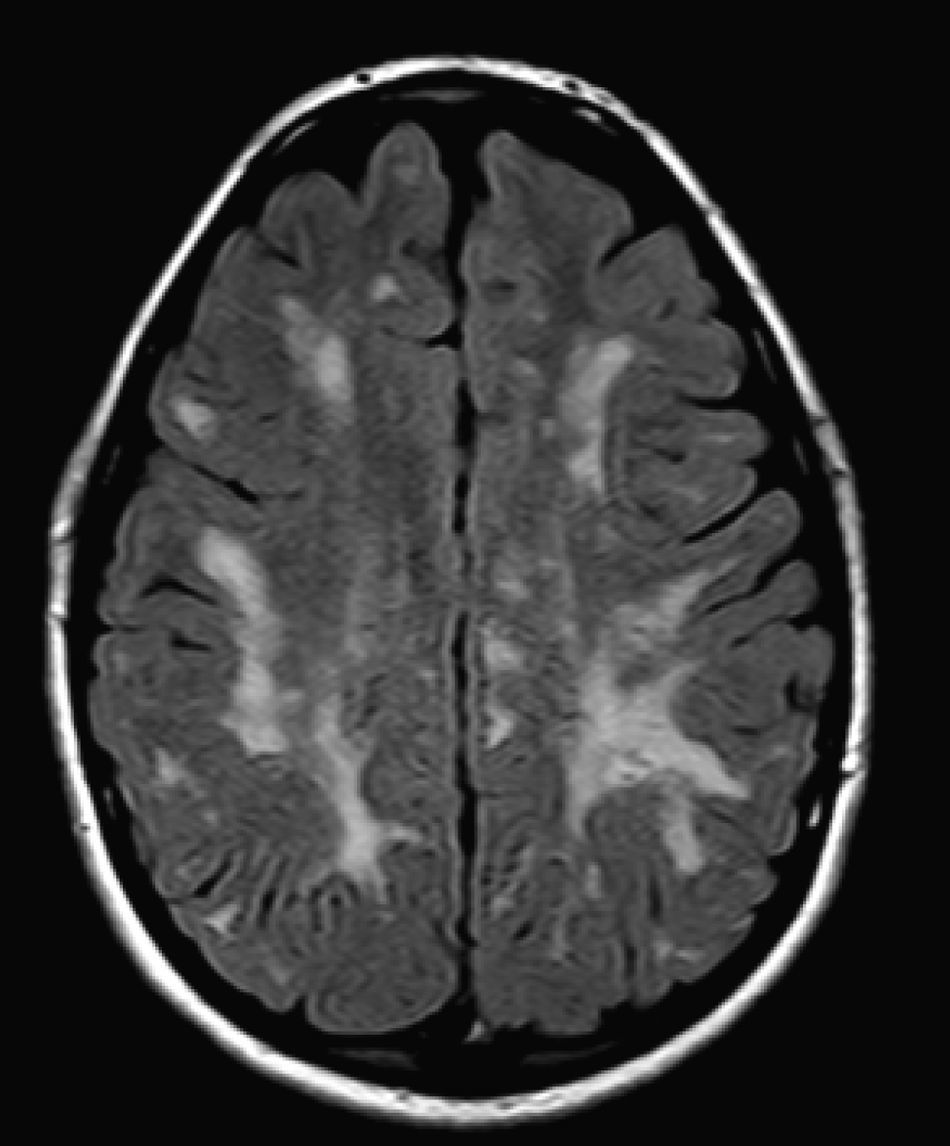
No encontramos una relación entre los hallazgos en la RMC y el tipo de MPS y la gravedad de la forma clínica. Si bien ninguna de las 2 niñas afectadas con el síndrome de Hurler-Scheie muestra las alteraciones parcheadas de la SB, uno de los 4 pacientes con Hurler tampoco las presenta. En 5 niños con Hunter moderado se encuentra un aumento de los EPVR, mientras que también se objetivan en otro con la forma grave de la enfermedad. Esto significaría que las alteraciones cerebrales descritas en nuestros pacientes (el aumento de los EPVR, la alteración parcheada de la SB, la ventriculomegalia, etc.) serían independientes de la gravedad. Pero hay estudios que sí hallan una relación directa de la severidad de estas alteraciones neurorradiológicas y la gravedad de la enfermedad13. Algunos en concreto encuentran relación entre el nivel cognitivo y la alteración de la SB (no con otras alteraciones en la neuroimagen)18. Otros relacionan la extensión de la afectación de la SB con la gravedad del deterioro cognitivo19. En nuestra serie contamos con un paciente con síndrome de Hunter moderado, que lleva más de 4 años con tratamiento enzimático sustitutivo semanal y seguimiento radiológico con RMC anual desde el diagnóstico hasta la actualidad. En el último control radiológico de este paciente se objetivó una progresión de las lesiones parcheadas en la SB (figs. 3 y 4); no obstante, el estudio neuropsicológico no mostró caídas significativas (superiores a 1 desviación típica) en el CI total, incluso reveló mejoría en áreas como procesamiento auditivo, lenguaje expresivo, memoria verbal, y menores dificultades motoras que las evaluadas en la exploración previa. Sin embargo, hay trabajos recientes en los que parece mostrarse una mejoría radiológica consecutiva al tratamiento enzimático sustitutivo (sobre todo de las lesiones de la SB) en pacientes con MPS tipo I atenuado20 y otros que documentan una mejoría o estabilización de las alteraciones en la neuroimagen en pacientes con MPS tipo I y II21.


Se ha descrito en MPS un aumento de partes blandas retroodontoideas, con disminución del calibre del conducto raquídeo (lo cual puede producir síntomas de compresión medular), así como otras alteraciones vertebrales1,14,22. En nuestra serie también encontramos alteraciones en la RM cervical y otras partes de la columna, lo que apoya la importancia de la realización de estas pruebas en pacientes con MPS, sobre todo si aparece clínica neurológica compatible.
Nuestra serie presenta claras limitaciones. Solo 5 pacientes cuentan con RMC seriada y la edad de realización de la RMC es heterogénea. Además, el número de pacientes es limitado por lo que los resultados obtenidos de esta serie son meramente descriptivos. Sería necesaria una muestra mucho más amplia para poder comparar y obtener resultados más fiables. En este grupo de enfermedades sería deseable poder evaluar los cambios neurorradiológicos evolutivos y estudiar la relación de estos hallazgos con el deterioro cognitivo. Probablemente, esto puede ser más fácil de ahora en adelante, dada la amplia disponibilidad de la RM. Las alteraciones radiológicas encontradas en las MPS no son específicas pero sí sugestivas de este grupo de enfermedades. Por tanto, ante un niño con hernias, limitación articular, visceromegalias, sordera, retraso mental, alteraciones óseas, o algún otro síntoma característico, y una imagen compatible en la RMC, se estudiará la posibilidad de una MPS.

