



Describir la historia del descubrimiento de la SCA36 y revisar los conocimientos actuales sobre esta entidad que, por un efecto fundador, ha pasado a ser la SCA más prevalente en Galicia (España).
DesarrolloLa SCA36 es una enfermedad heredodegenerativa autosómica dominante, de inicio tardío y lenta progresión, que cursa con ataxia cerebelosa, hipoacusia neurosensorial y discreta afectación de neuronas motoras (atrofia y fasciculaciones linguales y signos piramidales leves). Ha sido descrita inicialmente en Japón (ataxia del río Asida) y en Galicia (ataxia da Costa da Morte). Se produce por una mutación (expansión intrónica de hexanucleótido) en el gen NOP56, localizado en 20p13. En los estudios de resonancia magnética se observa inicialmente atrofia vermiana superior, que se extenderá al resto del cerebelo y finalmente a la porción bulboprotuberancial del tronco cerebral, sin lesiones de sustancia blanca. Las velocidades de conducción nerviosa periférica son normales y en los estudios de potenciales evocados somatosensoriales se detecta retraso de la conducción al estimular en miembros inferiores. En los pacientes con hipoacusia, suele encontrarse en la audiometría una caída>40dB a partir de 2.400Hz; también se observa ausencia de ondas i y ii en los estudios de potenciales evocados auditivos.
ConclusionesLa ataxia da Costa da Morte-SCA36 es la SCA más prevalente en Galicia (España). Dada la alta tasa de emigración de nuestra comunidad autónoma, se espera que se diagnostiquen nuevos casos en diversas latitudes, sobre todo en América Latina. Ahora está disponible el diagnóstico genético para pacientes con clínica y portadores asintomáticos. Dado el alto número de pacientes en riesgo de sufrir la enfermedad, continuamos con las investigaciones para aclarar los mecanismos moleculares patogénicos y poder encontrar una terapéutica.
To describe the history of the discovery of SCA36 and review knowledge of this entity, which is currently the most prevalent hereditary ataxia in Galicia (Spain) owing to a founder effect.
DevelopmentSCA36 is an autosomal dominant hereditary ataxia with late onset and slow progression. It presents with cerebellar ataxia, sensorineural hearing loss, and discrete motor neuron impairment (tongue atrophy with denervation, discrete pyramidal signs). SCA36 was first described in Japan (Asida River ataxia) and in Galicia(Costa da Morte ataxia). The condition is caused by a genetic mutation (intronic hexanucleotide repeat expansion) in the NOP56 gene on the short arm of chromosome 20 (20p13). Magnetic resonance image study initially shows cerebellar vermian atrophy that subsequently extends to the rest of the cerebellum and finally to the pontomedullary region of the brainstem without producing white matter lesions. Peripheral nerve conduction velocities are normal, and sensorimotor evoked potential studies show delayed conduction of stimuli to lower limbs. In patients with hearing loss, audiometric studies show a drop of >40dB in frequencies exceeding 2,500Hz. Auditory evoked potential studies may also show lack of waves I and II.
ConclusionsCosta da Morte ataxia or SCA36 is the most prevalent SCA in the Spanish region of Galicia. Given the region's history of high rates of emigration, new cases may be diagnosed in numerous countries, especially in Latin America. Genetic studies are now available to patients and asymptomatic carriers. Since many people are at risk for this disease, we will continue our investigations aimed at elucidating the underlying pathogenic molecular mechanisms and discovering effective treatment.
En el año 1864, Duchenne du Boulogne denominó «ataxia locomotriz» al trastorno de la marcha producido por la tabes dorsalis1. En 1893, Pierre Marie separaba las ataxias hereditarias dominantes de la ataxia recesiva, descrita ya anteriormente por Nikolaus Friedreich en 18632. A lo largo del siglo xx, Holmes, Dejérine, Greenfield y Thomas, entre otros autores, realizaron una clasificación de las ataxias degenerativas, basada en criterios clínicos y patológicos: ataxias cerebelosas, olivopontecerebelosas y espinocerebelosas, cada grupo subdividido en esporádicas y heredadas3–5. En el año 1980, Anita Harding6 clasificó las ataxias de herencia autosómica dominante (ADCA) en 3 grupos: ADCA I (síndrome cerebeloso plus, que puede acompañarse de oftalmoplejía, demencia, trastornos del movimiento, amiotrofia y neuritis óptica), ADCA II (síndrome cerebeloso asociado a degeneración retiniana) y ADCA III (síndrome cerebeloso puro). La clasificación de Harding conserva todavía una innegable utilidad clínica y es muy útil en la aproximación inicial ante un paciente con ataxia y patrón de herencia dominante.
Después de 1990, comienzan a identificarse los primeros loci cromosómicos de algunas ADCA7,8 y este acrónimo da paso al de SCA (ataxias espinocerebelosas, del inglés spinocerebellar ataxias), que se utiliza actualmente para designar a un grupo heterogéneo y cada vez más extenso de trastornos degenerativos, que tienen en común el incluir ataxia entre sus manifestaciones clínicas relevantes y heredarse siguiendo un patrón autosómico dominante. A pesar de que se han enumerado 40 SCA, por el momento solo se tiene certeza de 34 loci cromosómicos, ya que la SCA9 es una incógnita, la 15 y la 16 son la misma, la 24 tiene patrón de herencia recesivo, y la 33 no tiene soporte bibliográfico que la avale9–13.
La heterogeneidad clínica de las SCA se corresponde en cierto modo con el tipo alteración genética que las causa. Así los tipos de mutaciones conocidos hasta la actualidad son: 1) mutaciones puntuales (SCA13, 23, 27, 35, 38, 40); 2) mutaciones con desplazamiento del marco de lectura (SCA11); 3) deleciones (SCA15, 27); 4) duplicaciones (SCA20); 5) repeticiones-expansiones de poliglutamina (tripletes de CAG) en regiones codificantes (SCA1, 2, 3, 7, 17 y DRPLA); 6) repeticiones-expansiones de tripletes, quintupletes y hexapletes en regiones no codificantes (SCA8, 10, 12, 31 y 36). En algunas SCA (SCA4, 10, 18, 19, 21, 22, 25, 26, 29, 32, 34, 37) solo se dispone de un estudio de ligamiento en una región cromosómica determinada pero se desconoce el gen y el tipo de mutación9–13.
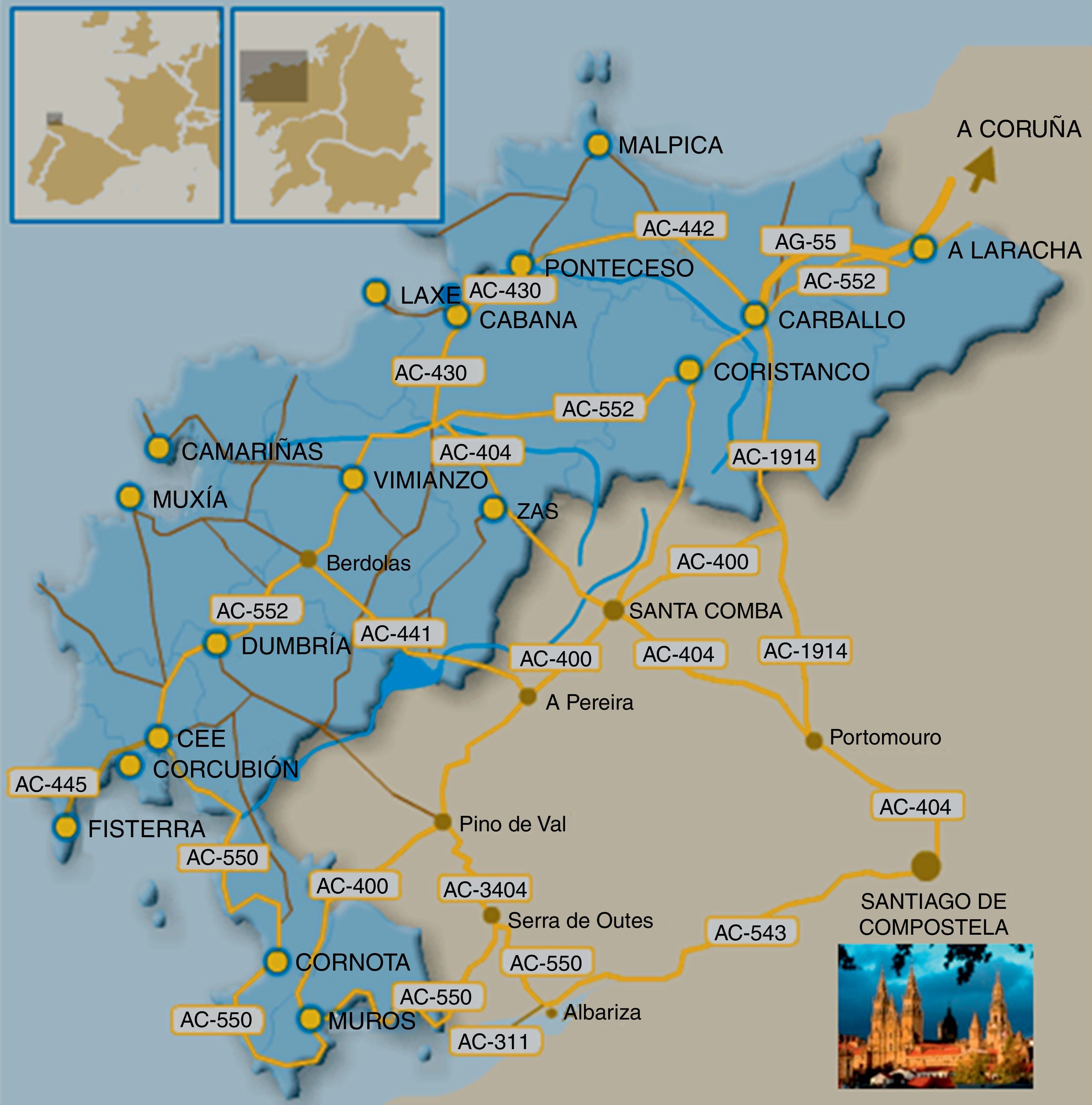
En el presente trabajo revisamos la SCA36, la última SCA descrita en la que se conoce la mutación genética, y que hemos denominado «SCA da Costa Morte», ya que en dicha comarca atlántica gallega (fig. 1) hemos estudiado inicialmente a 2 largas familias con un importante número de afectados (más de 40) y portadores (más de 60), hecho que condiciona el que hoy por hoy sea el tipo de SCA más prevalente en Galicia (España), que al ser tierra de emigrantes condiciona que la enfermedad pueda estar bastante extendida.
DesarrolloHistoria del descubrimiento
Desde los inicios de la década de los pasados 90, consultaron en el Complexo Hospitalario Universitario de Santiago diversos pacientes procedentes de los municipios de Muxia y Ponteceso, pertenecientes a la comarca de A Costa da Morte (A Coruña-España) (fig. 1). Todos ellos presentaban un cuadro de ataxia de inicio relativamente tardío y lenta progresión, y manifestaban que en su entorno familiar había casos similares; por los datos suministrados, el patrón de herencia era autosómico dominante. A medida que los estudios genéticos de las primeras SCA fueron estando disponibles, fuimos comprobando que estos enfermos no eran portadores de las mutaciones o ligamientos genéticos que se iban describiendo. En el año 2005 ya teníamos asumido que estábamos ante una enfermedad relativamente homogénea, con muchos pacientes concentrados en la mencionada área geográfica, por lo que decidimos realizar un amplio estudio de campo (colaboración de neurólogos y genetistas), visitando a los pacientes y a los familiares en riesgo en su medio, a la vez que, tras firmar el pertinente consentimiento informado, se obtuvieron muestras para los estudios genéticos y se confeccionó un extenso árbol genealógico. En las Reuniones de la Sociedade Galega de Neuroloxía (A Coruña, 2008) y de la Sociedad Española de Neurología (Barcelona, 2008) Arias et al.14 comunicaron las características fundamentales del cuadro clínico de lo que pensamos era una nueva SCA. En el año 2011 un grupo japonés (Kobayashi et al.)15 publicó por vez primera la mutación de la SCA36 y poco después lo hizo nuestro grupo (García-Murias et al.)16. En la actualidad ya se han comunicado otros casos en España, Italia y Polonia.
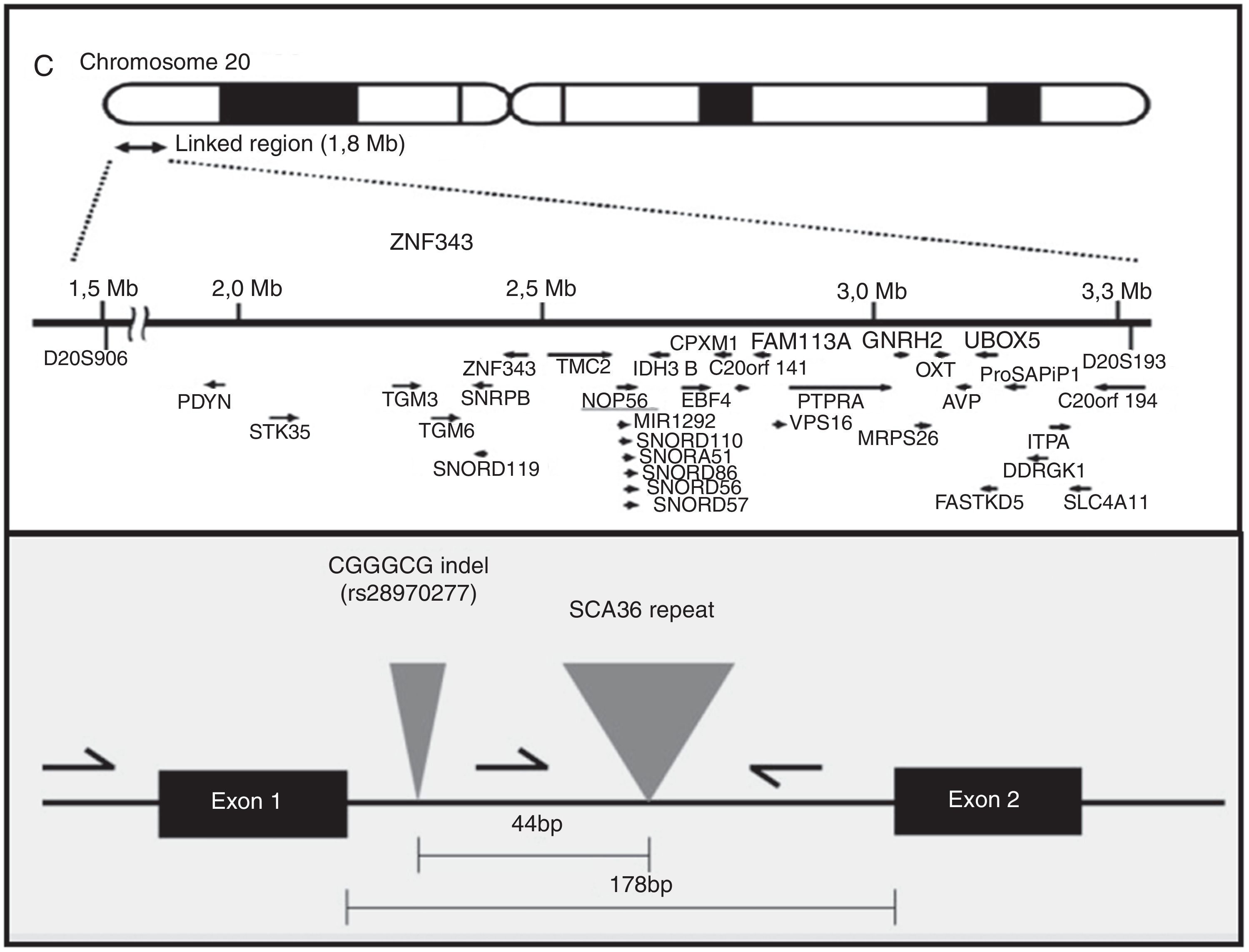
EtiopatogeniaEl gen NOP56, localizado en 20p13, codifica una proteína de 56kD, que interactúa con NOP1 y NOP58 para formar la subunidad ribosómica de 60S. La SCA36 está causada por una mutación en el gen NOP56, en concreto por una expansión en heterocigosis del polimorfismo GGCCTG, localizada en el intrón 1 (fig. 2). Se ha comprobado en estudios con células linfoblásticas que la expansión intrónica de NOP56 da lugar a acumulaciones focales intranucleares de ácidos ribonucleicos, que podrían interferir con diversos factores de transcripción15. Algo similar sucede en otras SCA producidas por expansiones, y también en la enfermedad de Huntington y en la distrofia miotónica. El papel del ácido ribonucleico en las enfermedades neurodegenerativas es actualmente un campo de creciente interés17.
Epidemiología
La prevalencia de las SCA varía considerablemente de unos países a otros; en términos generales podemos decir que la SCA3 es la más frecuente en el planeta, pero en México lo es la SCA10, en los países escandinavos la SCA7, y en España la SCA2 y la SCA3 tienen una frecuencia similar18–21. En Galicia, la SCA36 es ahora la más prevalente y representa el 21,3% de las ataxias del adulto con patrón de herencia dominante, según la base de datos de la Fundación Pública Gallega de Medicina Xenómica16. La relación entre los ancestros gallego y japonés así como otros casos puntuales detectados en España y Europa está por aclarar. En todo caso, dado que la emigración, dirigida tanto hacia el resto del Estado Español como a Europa y América Latina, es un fenómeno muy ligado a Galicia, es de esperar que pronto se detecten nuevos casos en tales lugares.
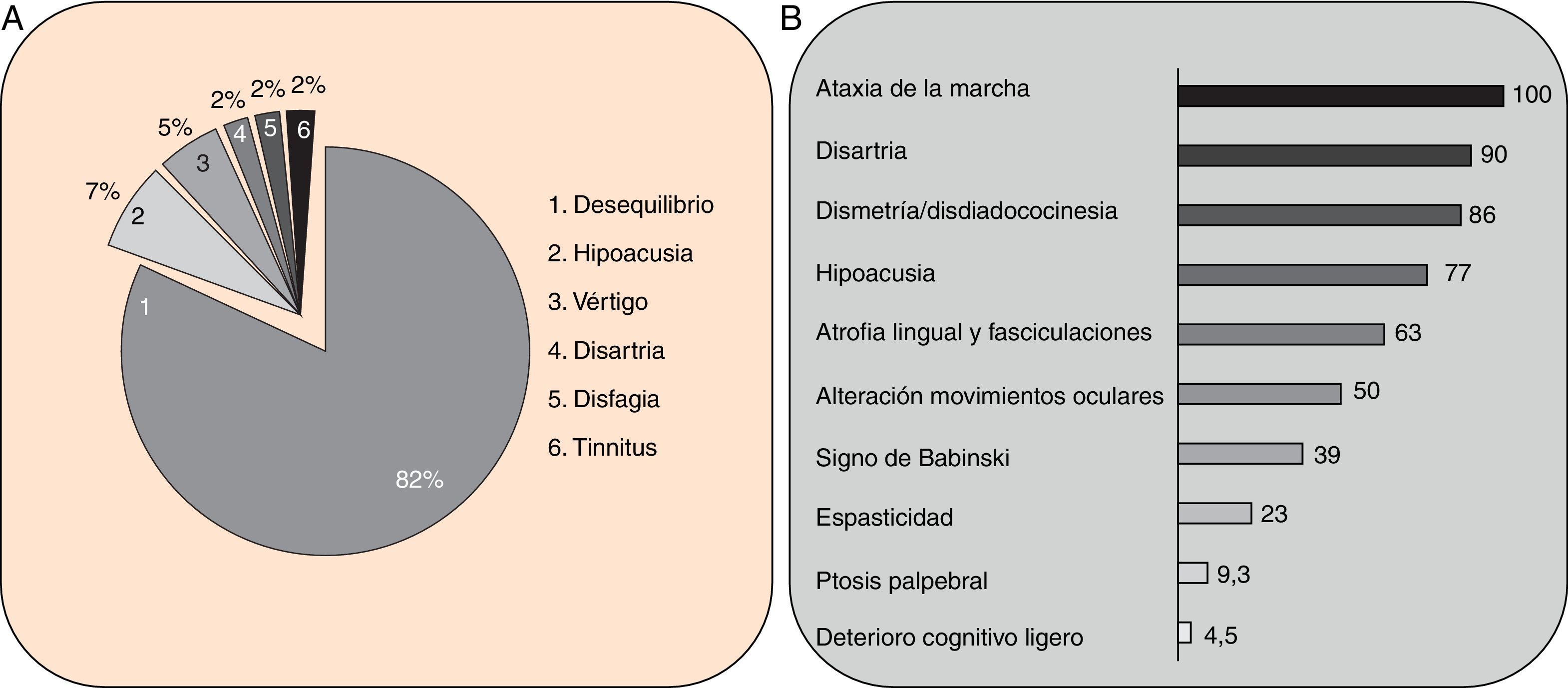
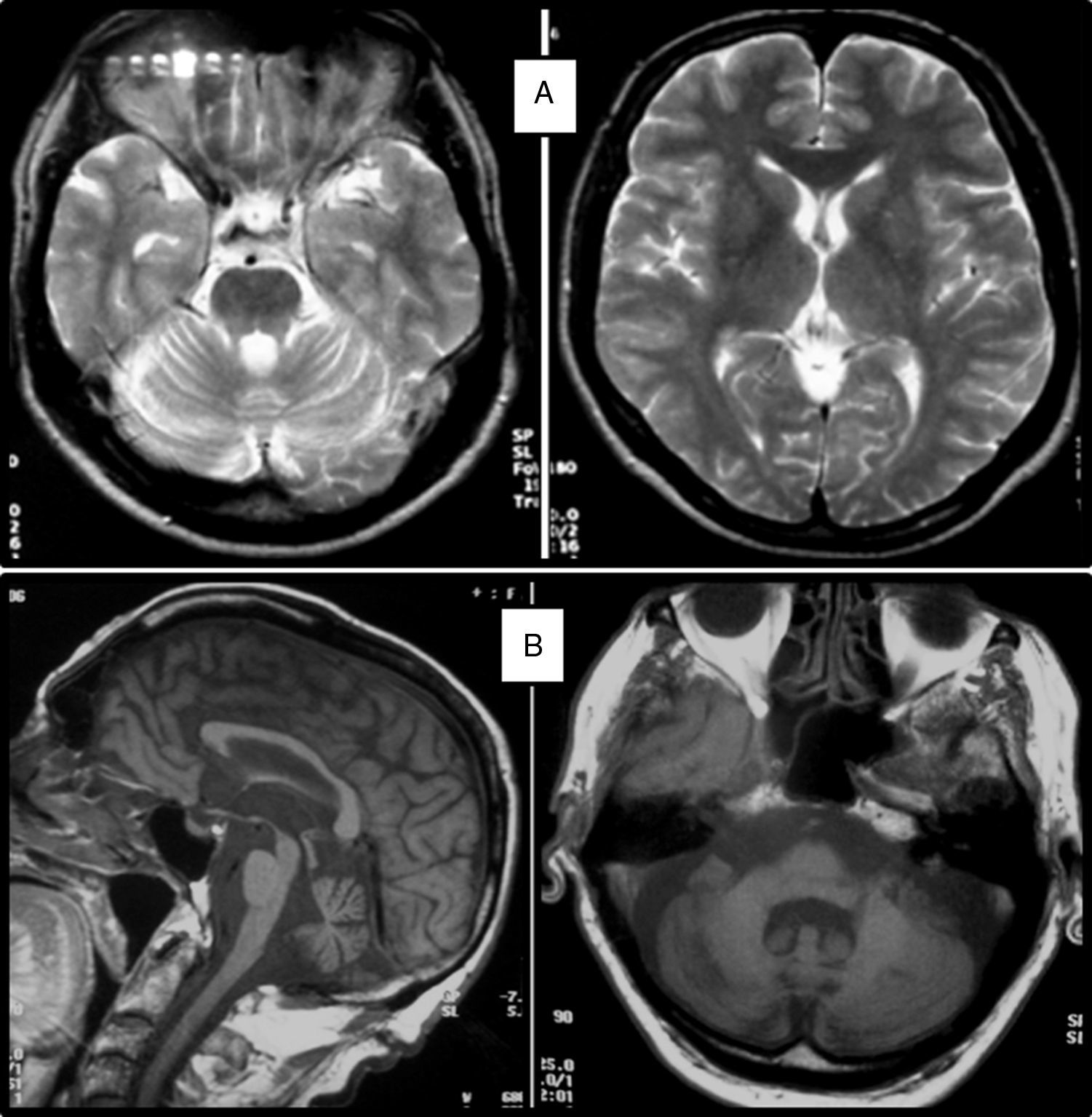
Cuadro clínicoUn cuadro de desequilibrio e inestabilidad, de inicio entre los 40 y 60 años de edad, suele ser el síntoma inicial de la SCA36. Este cuadro suele progresar lentamente y a él se le unirá ataxia apendicular con dismetría y disdiacocinesia, además de disartria de características cerebelosas o mixta (cerebelosa y bulbar)14-16,22,23. Raramente los enfermos llegan a precisar silla de ruedas antes de los 15 años de evolución. Alrededor del 80% de los pacientes presentan hipoacusia sensorial, afectación no mencionada en las primeras publicaciones de pacientes japoneses, aunque reconocida posteriormente15,24. En el 60-70% de los pacientes hemos detectado atrofia de la lengua con fasciculaciones, que puede contribuir a la mencionada disartria y, en pocos casos, a disfagia leve o moderada, ya que ningún paciente ha precisado la colocación de sonda nasogástrica o gastrostomía percutánea para asegurar su alimentación15,16,22–26. Nosotros no hemos encontrado otros síntomas o signos de enfermedad de motoneurona inferior fuera de la lengua, aunque sí se han notificado en algunos casos nipones. En más de la mitad de los casos está presente un cuadro piramidal con hiperreflexia y signo de Babinski con escasa o nula espasticidad. Otros signos clínicos posibles son nistagmo, sacadas lentas e hipométricas, ptosis palpebral y deterioro cognitivo ligero o moderado con un patrón de síndrome disejecutivo frontal25. En la figura 3 se detallan los porcentajes de los síntomas de comienzo y de las manifestaciones integrantes del cuadro clínico, correspondientes a nuestra serie inicial de 44 pacientes. No hemos encontrado clínica de disautonomía o alteraciones extrapiramidales, aunque Miyashiro et al.27 han comunicado recientemente un caso que asociaba parkinsonismo y distonía. Las aportaciones más relevantes de los medios complementarios en la SCA3615,16 se resumen en: A) En los estudios de resonancia magnética (RM) se observa atrofia cerebelosa desde el inicio de los síntomas; suele comenzar en el vermis superior y se extiende al resto del cerebelo para finalmente afectar al tronco cerebral y conformar un patrón de atrofia olivopontocerebelosa (fig. 4); no se detectan lesiones de sustancia blanca; en algunos casos puede ser evidente una moderada atrofia frontal. B) En los estudios neurofisiológicos se hallan valores normales para las velocidades de conducción motora y sensitiva; la denervación se restringe a la lengua en los pacientes gallegos. En el estudio de potenciales evocados somatosensoriales se encuentran retrasos de la conducción al estimular en miembros inferiores. El estudio de los potenciales evocados auditivos muestra ausencia o marcada reducción de amplitud de las ondas i y ii. En la audiometría es características una caída de 40 o más dB en frecuencias superiores a 2.500Hz. C) En los escasos estudios histopatológicos realizados se encontró pérdida de células de Purkinje, sobre todo en el núcleo dentado del cerebelo, y pérdida de neuronas motoras en el núcleo del hipogloso. No se observaron, tal como sucede en la ELA, inclusiones citoplasmáticas eosinofílicas tipo Bunina en motoneuronas del asta anterior medular.
 Porcentaje de pacientes de SCA36-ACM que presentaron como síntoma inicial los señalados. B) Porcentaje de pacientes de SCA36-ACM que presentaban en el examen clínico las manifestaciones clínicas señaladas.")
 Estudio de RM-T2 en un paciente con SCA-ACM de 52 años: ausencia de lesiones de sustancia blanca y atrofia cortical; atrofia cerebelosa difusa. B) Estudio de RM-T1 en un paciente con SCA-ACM de 86 años: patrón de atrofia olivopontocerebelosa.")
No hemos encontrado una clara correlación entre adelanto y gravedad de las manifestaciones clínicas y el tamaño de la expansión14.
Diagnóstico diferencialEn sus inicios, la SCA36 tiene el perfil de una ADCA tipo iii (síndrome cerebeloso puro), más adelante con la hipoacusia y las fasciculaciones linguales podríamos considerarla una ADCA tipo ilight. Por lo tanto, al principio el diagnóstico diferencial deberá hacerse con las SCA5, 16, 11, 26, 30 y 31. En general, puede afirmarse que las ADCA tipo i (SCA1, 2, 3) tienen todas un comienzo más precoz9–13. Si nos centramos en la hipoacusia, esta manifestación también está presente en la SCA31 y ocasionalmente en otras28. Por lo que respecta a la afectación de neuronas motoras, también puede encontrarse en la SCA229 y en la atrofia dentato-rubro-pálido-luysiana (DRPL). El síndrome de temblor y ataxia asociado a X-frágil podría tener cierta similitud clínica con la SCA36, aunque el estudio de RM suele mostrar en esta entidad lesiones hiperintensas características en los pedúnculos cerebelosos medios30.
La ataxia de Friedreich y otras ataxias recesivas como la ataxia con apraxia oculomotora tipo 1 y 2, la ataxia telangiectasia y la xantomatosis cerebrotendinosa pueden excepcionalmente tener un comienzo tardío y cursar con un fenotipo suavizado que recuerde al de la SCA36, aunque suelen incluir datos clínicos o neurofisiológicos de neuropatía periférica31; por lo tanto deberán tenerse en cuenta ante casos aislados. Las citopatías mitocondriales, tanto las derivadas de mutaciones del ADN mitocondrial y por lo tanto de transmisión materna como las derivadas de mutaciones del ADN somático32, cursan con un amplio rango de síntomas, entre ellos ataxia e hipoacusia, y por lo tanto podrían confundirse con los de la SCA36. Las variedades de inicio cerebeloso de la atrofia multisistema y de la parálisis supranuclear progresiva también pueden remedar a la SCA36 en sus inicios, pero la evolución y los datos de RM serán netamente diferentes.
Un importante número de ataxias adquiridas de inicio tardío deben considerarse también en el diagnóstico de casos aislados de SCA36: tóxicas (alcoholismo, drogas), metabólicas (deficiencia de vitamina E y encefalopatía de Wernicke), autoinmunes (síndrome de Miller Fisher, encefalopatía de Bickerstaff, enfermedad celíaca, anti-GAD y otros anticuerpos) y paraneoplásicas33.
Diagnóstico y consejo genéticoLa identificación o no de la expansión patológica del hexanucleótido GGCCTG en el gen NOP56 confirmará o descartará el diagnóstico de SCA36 en un sujeto en riesgo de padecerla o en un caso aparentemente esporádico de ataxia. Mediante PCR y los adecuados primers se detecta la expansión en heterocigosis; para cuantificar con exactitud el tamaño de un alelo con una gran expansión hay que recurrir al análisis de southern blot, que habitualmente no es necesario. Los alelos normales tienen repeticiones de CGCCTG que varían entre 3 y 1416,34 en los controles caucasianos y de 3 a 8 en los japoneses15. La expansión del alelo mutado tiene 650 o más repeticiones. El diagnóstico en sujetos asintomáticos y el diagnóstico preimplantación y prenatal requieren identificar la mutación en la familia. Hasta la fecha no se conocen otros trastornos alélicos con un fenotipo distinto por mutaciones en NOP56.
TratamientoLa SCA36 carece, por el momento, de tratamiento específico. En estos momentos los pacientes y los portadores de la mutación (que han querido voluntariamente conocer su status genético) superan ampliamente, en Galicia, el centenar. A este colectivo debemos recomendarle ejercicio físico regular, evitar ganancia ponderal y consumo de alcohol y de otras sustancias y medicación potencialmente tóxica para el cerebelo (fenitoína, carbamazepina, metronidazol, amiodarona, litio) y el sistema auditivo (salicilatos). También evitarán el trauma acústico utilizando auriculares si están expuestos a ruido ambiental excesivo en su actividad laboral.
Investigaciones futurasPara comprender los mecanismos moleculares de la SCA36 e identificar potenciales estrategias terapéuticas nos planteamos ahora 3 bloques de cuestiones: epidemiología y filogenética de la mutación, dinámica de la expansión, inestabilidad mitótica y meiótica, influencia en las características fenotípicas y efectos sobre la transcripción de otros genes. Esperamos que todas estas investigaciones, que ya hemos puesto en marcha, abran las puertas para una terapéutica eficaz, al menos para los numerosos portadores por el momento asintomáticos y los mínimamente afectados.
ConclusionesDescubierta su mutación causal (expansión intrónica de hexanucleótido en el gen NOP56), la ataxia da Costa da Morte se incluye ahora dentro de la SCA36. Su fenotipo lo conforman un síndrome cerebeloso de inicio tardío y lenta progresión, asociado a hipoacusia neurosensorial, atrofia y fasciculaciones linguales, y discretos signos piramidales. Esta entidad es la SCA más prevalente en Galicia. Actualmente ya es posible el diagnóstico en sujetos portadores y realizar consejo genético con vistas a reproducción. Está por aclarar la relación (¿efecto fundador independiente o común?) de la ataxia da Costa da Morte con la ataxia del río Asida, descrita en Japón y producida por una mutación similar.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

