



La distrofia muscular de Duchenne (DMD) es una enfermedad neuromuscular grave que afecta a uno de cada 3.500 varones nacidos y sigue un patrón de herencia ligada al cromosoma X. En esta enfermedad se observa una ausencia total de la distrofina, generalmente debida a mutaciones en el gen DMD, que altera la pauta de lectura y en torno al 80% de los casos son debidos a deleciones y duplicaciones de uno o más exones.
MétodosSe han revisado 284 casos de varones diagnosticados genéticamente de DMD entre los años 2007 y 2014. Estos pacientes provienen de 8 hospitales españoles de referencia que cubren la mayor parte del territorio español. Para la identificación de las mutaciones se realizaron las técnicas de reacción en cadena de la polimerasa multiplex, MLPA y secuenciación.
ResultadosLos pacientes con DMD presentan en su mayoría grandes deleciones (46,1%) o grandes duplicaciones (19,7%) en el gen de la distrofina. El restante 34,2% corresponde al conjunto de mutaciones puntuales, destacando las sustituciones nucleotídicas tipo nonsense que aparecen en la mitad de los casos. Este estudio permitió identificar 23 nuevas mutaciones en DMD: 7 grandes deleciones y 16 mutaciones puntuales.
ConclusionesEl algoritmo de diagnóstico genético aplicado por los centros participantes es el más adecuado para genotipificar a los pacientes con DMD. La especificidad genética de las distintas terapias en desarrollo pone de manifiesto la importancia de conocer la mutación de cada paciente, siendo un 38,7% de ellos susceptibles de participar en los ensayos clínicos actuales.
Duchenne muscular dystrophy (DMD) is a severe X-linked recessive neuromuscular disease that affects one in 3500 live-born males. The total absence of dystrophin observed in DMD patients is generally caused by mutations that disrupt the reading frame of the DMD gene, and about 80% of cases harbour deletions or duplications of one or more exons.
MethodsWe reviewed 284 cases of males with a genetic diagnosis of DMD between 2007 and 2014. These patients were selected from 8 Spanish reference hospitals representing most areas of Spain. Multiplex PCR, MLPA, and sequencing were performed to identify mutations.
ResultsMost of these DMD patients present large deletions (46.1%) or large duplications (19.7%) in the dystrophin gene. The remaining 34.2% correspond to point mutations, and half of these correspond to nonsense mutations. In this study we identified 23 new mutations in DMD: 7 large deletions and 16 point mutations.
ConclusionsThe algorithm for genetic diagnosis applied by the participating centres is the most appropriate for genotyping patients with DMD. The genetic specificity of different therapies currently being developed emphasises the importance of identifying the mutation appearing in each patient; 38.7% of the cases in this series are eligible to participate in current clinical trials.
Las distrofinopatías constituyen un grupo de enfermedades neuromusculares causadas por alteraciones en el gen de la distrofina (DMD). Este gen, uno de los más grandes encontrados en humanos (79 exones y aproximadamente 2,2Mb de ADN genómico1), se localiza en la región cromosómica Xp21, por lo que son enfermedades que siguen un patrón de herencia ligada al cromosoma X. La distrofina es una proteína muscular de 427kDa que desempeña un papel fundamental en la estabilización del sarcolema con el anclaje, a través del complejo de glucoproteínas asociadas a la distrofina, de los filamentos de actina en el citoesqueleto y la matriz extracelular. La ausencia de distrofina da lugar a la rotura de esta conexión provocando alteraciones en la membrana plasmática y, finalmente, la degeneración y la necrosis de las miofibras2. De forma esquemática, las mutaciones en la distrofina dan lugar a 2 formas clínicas fundamentales de distrofia muscular: una forma grave, la distrofia muscular de Duchenne (DMD; MIM #310200), y otra más leve, la distrofia muscular de Becker (DMB; MIM#300376). Otras distrofinopatías incluyen los denominados «outliers» o formas intermedias, el síndrome de mialgia y calambres y la miocardiopatía dilatada ligada a X. Con menor frecuencia, las mujeres portadoras pueden ser sintomáticas, con cuadros de gravedad variable3-5.
La DMD es la enfermedad muscular hereditaria más frecuente, con una incidencia estimada de 1 por cada 3.500 varones nacidos6. En esta enfermedad se observa una ausencia total de distrofina, generalmente debida a mutaciones que alteran la pauta de lectura dando lugar a una proteína truncada y no funcional. Estas mutaciones suelen generar un codón de parada prematuro por lo que se activa el mecanismo de degradación de transcritos alterados nonsense-mediated mRNA decay (NMD)7. Por otra parte, la DMB y otras distrofinopatías son causadas mayoritariamente por mutaciones que no alteran la pauta de lectura de la distrofina, por lo que se genera una proteína semifuncional. Esta correlación fenotipo-genotipo explica el 92% de los casos y se conoce como la hipótesis de la alteración de la pauta de lectura8.
En la DMD se han descrito diversos tipos de mutaciones; alrededor del 65-70% de los casos son debidos a grandes deleciones (de uno o más exones) y en torno al 7-10% a grandes duplicaciones (de uno o más exones). El resto de los casos con DMD/BMD se debe a mutaciones puntuales (principalmente nonsense) y pequeñas deleciones e inserciones9-12.
A lo largo de los años, se han publicado diversos estudios epidemiológicos que muestran datos de incidencia y prevalencia de DMD en distintos grupos poblacionales: Suecia13; Dinamarca14; norte de Inglaterra15; Irlanda del Norte16; noroeste de Toscana17; Estonia18; Norteamérica19; Sudáfrica20; Egipto21; China22, y Japón23. Además, en 2014 se publica una revisión sistemática y metaanálisis sobre la epidemiología de Duchenne, donde se estima una prevalencia global de 4,78 por cada 100.000 varones24.
Los pacientes con enfermedad de Duchenne son diagnosticados habitualmente entre los 3-5 años de edad y cursan con debilidad muscular progresiva y pérdida de la deambulación sobre los 13 años de edad. En aproximadamente un 30% de los casos se asocia a coeficiente intelectual bajo25. También es común la afectación cardíaca y respiratoria, que normalmente conlleva a la muerte entre la segunda y la tercera décadas de vida26.
En este artículo se recoge la información relativa a las alteraciones genéticas que subyacen en un amplio grupo de pacientes diagnosticados de enfermedad de Duchenne de la población española entre los años 2007 y 2014, permitiendo realizar una visión global del estado de esta enfermedad en España y revisar su algoritmo de diagnóstico genético.
Pacientes y métodosSe han revisado un total de 284 casos de varones diagnosticados genéticamente de DMD entre enero del 2007 y diciembre del 2014. Provienen de 8 hospitales españoles de referencia que cubren la mayor parte del territorio español: 151 casos del Hospital de la Santa Creu i Sant Pau (Barcelona), 39 casos del Hospital Universitario Virgen del Rocío (Sevilla), 38 casos del Hospital Universitari i Politècnic La Fe (Valencia), 26 casos del Hospital Universitario de la Paz (Madrid), 18 casos del Hospital Universitario Fundación Jiménez Díaz (Madrid) y 12 casos recogidos entre el Complexo Hospitalario Universitario de Vigo (Pontevedra), Hospital Universitario de Basurto (Vizcaya) y Hospital Clínico San Carlos (Madrid). El diagnóstico de DMD fue establecido por diversos especialistas basándose en estrictos criterios que incluyen: presentación clínica indicativa de DMD, antecedentes familiares de distrofia muscular de herencia ligada al cromosoma X, análisis genético del gen DMD y/o estudio de la biopsia muscular con análisis de distrofina mediante inmunohistoquímica o Western blot.
Extracción de ADNSe realizó la extracción del ADN genómico de los pacientes a partir de leucocitos de sangre periférica mediante protocolos estandarizados. Todos los pacientes o tutores, en el caso de menores de edad, firmaron el consentimiento informado para la realización de estudios genéticos.
Reacción en cadena de la polimerasa multiplexLa reacción en cadena de la polimerasa (PCR) multiplex permite la identificación de grandes deleciones en DMD mediante la amplificación simultánea, sobre la base de 2 series de oligonucleótidos27,28, del promotor muscular y 17 exones que cubren las regiones donde se acumula el mayor número de deleciones de la distrofina (Pm, 3, 4, 6, 8, 12, 13, 17, 19, 43, 44, 45, 47, 48, 50, 51, 52 y 60). Algunos laboratorios de referencia complementaron la PCR multiplex con una tercera serie de oligonucleótidos que incluye el promotor cerebral y 6 exones más de la distrofina (2, 20, 33, 53, 54 y 66).
Multiplex ligation-dependent probe amplificationEL análisis por multiple ligation-dependent probe amplification (MLPA) permite identificar tanto grandes deleciones como duplicaciones en DMD mediante hibridación de sondas específicas marcadas y amplificadas por PCR. En aquellos pacientes en los que no se identificó una deleción en el gen de la distrofina por PCR multiplex, o incluso como primera aproximación genética, se realizó un análisis de deleciones/duplicaciones mediante esta técnica utilizando los kits SALSA P034 y P035, siguiendo las instrucciones establecidas por el fabricante (MRC-Holland, Amsterdam).
SecuenciaciónLa detección de mutaciones puntuales se realizó mediante secuenciación directa basada en la técnica de Sanger de los 79 exones y las regiones intrónicas flanqueantes del gen DMD a partir de ADN genómico. En aquellos casos en los que se disponía de tejido muscular, el análisis molecular se realizó en primer lugar en ADNc retrotranscrito a partir del ARN mensajero extraído de biopsia muscular y las mutaciones identificadas fueron confirmadas posteriormente en el ADN genómico.
En algunos centros se realizó el análisis de mutaciones puntuales mediante la secuenciación de nueva generación en la plataforma MiSeq de Illumina o en la plataforma PGM de Life Technologies.
Confirmación de mutacionesSe actualizaron todas las mutaciones recogidas en el estudio siguiendo las normas establecidas por la Human Genome Variation Society (HGVS, www.hgvs.org), la Leiden Open Variant Database (LOVD, www.dmd.nl) y el Exome Variant Server (http://evs.gs.washington.edu/EVS/) a fin de confirmar su nomenclatura y determinar si habían sido descritas previamente. Las posiciones nucleotídicas se determinaron en relación con la secuencia de referencia de DMD (RefSeq NM_004006.2). La evaluación de la patogenicidad de las nuevas variaciones identificadas fue realizada en base a análisis «in silico» mediante algoritmos computacionales de predicción. La alteración de los sitios de splicing fue analizada mediante diferentes matrices de peso posicional integradas en los programas Splicing Finder y Alamut. En cuanto a las alteraciones por sustituciones aminoacídicas, fueron valoradas mediante los programas Polyphen-2 y SIFT.
Análisis de datos poblacionalesPara los estudios de prevalencia, a partir de los datos obtenidos, se utilizó la información recogida por el Instituto Nacional de Estadística teniendo en cuenta el 1 de enero del 2015 como punto de referencia para los cálculos.
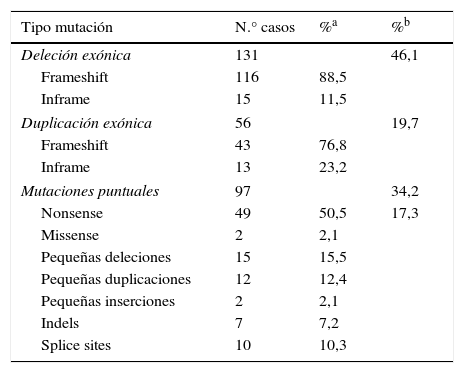
ResultadosGrandes deleciones y duplicacionesCon el análisis mutacional de DMD en los 284 pacientes, combinando las técnicas de PCR multiplex y MLPA, se encontraron 187 pacientes que presentaban deleciones o duplicaciones de uno o más exones, correspondiendo a un 46,1% (131/284) y un 19,7% (56/284), respectivamente (tabla 1).
Mutaciones identificadas en DMD
| Tipo mutación | N.° casos | %a | %b |
|---|---|---|---|
| Deleción exónica | 131 | 46,1 | |
| Frameshift | 116 | 88,5 | |
| Inframe | 15 | 11,5 | |
| Duplicación exónica | 56 | 19,7 | |
| Frameshift | 43 | 76,8 | |
| Inframe | 13 | 23,2 | |
| Mutaciones puntuales | 97 | 34,2 | |
| Nonsense | 49 | 50,5 | 17,3 |
| Missense | 2 | 2,1 | |
| Pequeñas deleciones | 15 | 15,5 | |
| Pequeñas duplicaciones | 12 | 12,4 | |
| Pequeñas inserciones | 2 | 2,1 | |
| Indels | 7 | 7,2 | |
| Splice sites | 10 | 10,3 | |
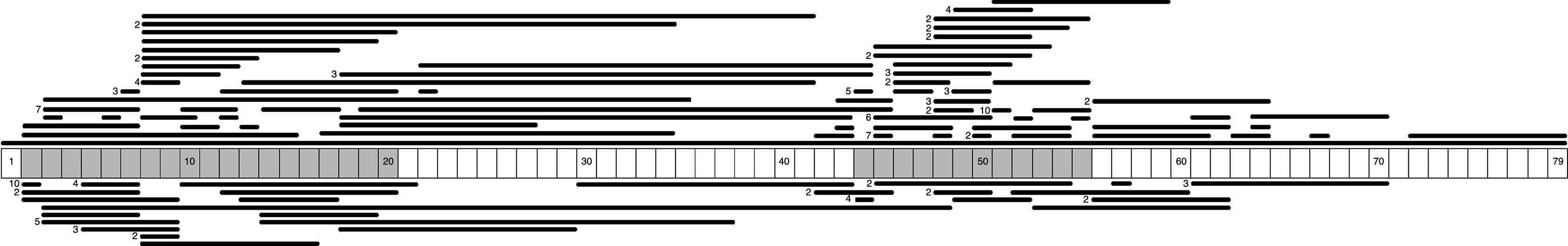
Se identificaron 71 tipos distintos de grandes deleciones, siendo la deleción del exón 51 la más frecuente (10/131: 7,6%), seguida de la deleción del exón 45 (7/131: 5,3%), las deleciones de los exones 3-7 y 45-50 en igual frecuencia (6/131: 4,6%), y la deleción del exón 44 (5/131: 3,8%). Tras el análisis de la longitud de las grandes deleciones identificadas, se observó que las más frecuentes eran aquellas que implican a un único exón, presentes en un 29% (38/131) de los casos con grandes deleciones. A partir de estos datos, junto a la distribución del resto de grandes deleciones identificadas en el estudio, se confirma la clusterización preferente en 2 regiones del gen de la distrofina: la región próxima al 5′ del dominio N-terminal (exones 2 al 20) y la región media-distal del dominio central (exones 44 al 55) con un 32,7% y 57,4%, respectivamente, correspondientes a las denominadas zonas calientes del gen o «hot spots» (fig. 1).

Respecto al efecto a nivel de transcrito de las grandes deleciones, el 88,5% (116/131) de las identificadas en nuestro estudio rompieron la pauta de lectura, mientras que un 11,5% (15/131) de casos presentaron deleciones que mantenían la pauta de lectura, por lo que no cumplían la hipótesis de Monaco8. En estos casos, el diagnóstico de DMD se basó en criterios clínicos y/o referentes a la biopsia muscular (tabla 1).
Se identificaron 27 tipos distintos de grandes duplicaciones, siendo la más frecuente la duplicación del exón 2 (10/56: 17,9%). Al igual que en el caso de las grandes deleciones, se observó que las duplicaciones más frecuentes eran aquellas que implican a un único exón (15/56: 26,8%). Analizando la localización de las grandes duplicaciones dentro de la proteína, también se observó un agrupamiento en la región próxima al 5′ del dominio N-terminal (67,9%), y en la región media-distal del dominio central (28,6%) (fig. 1).
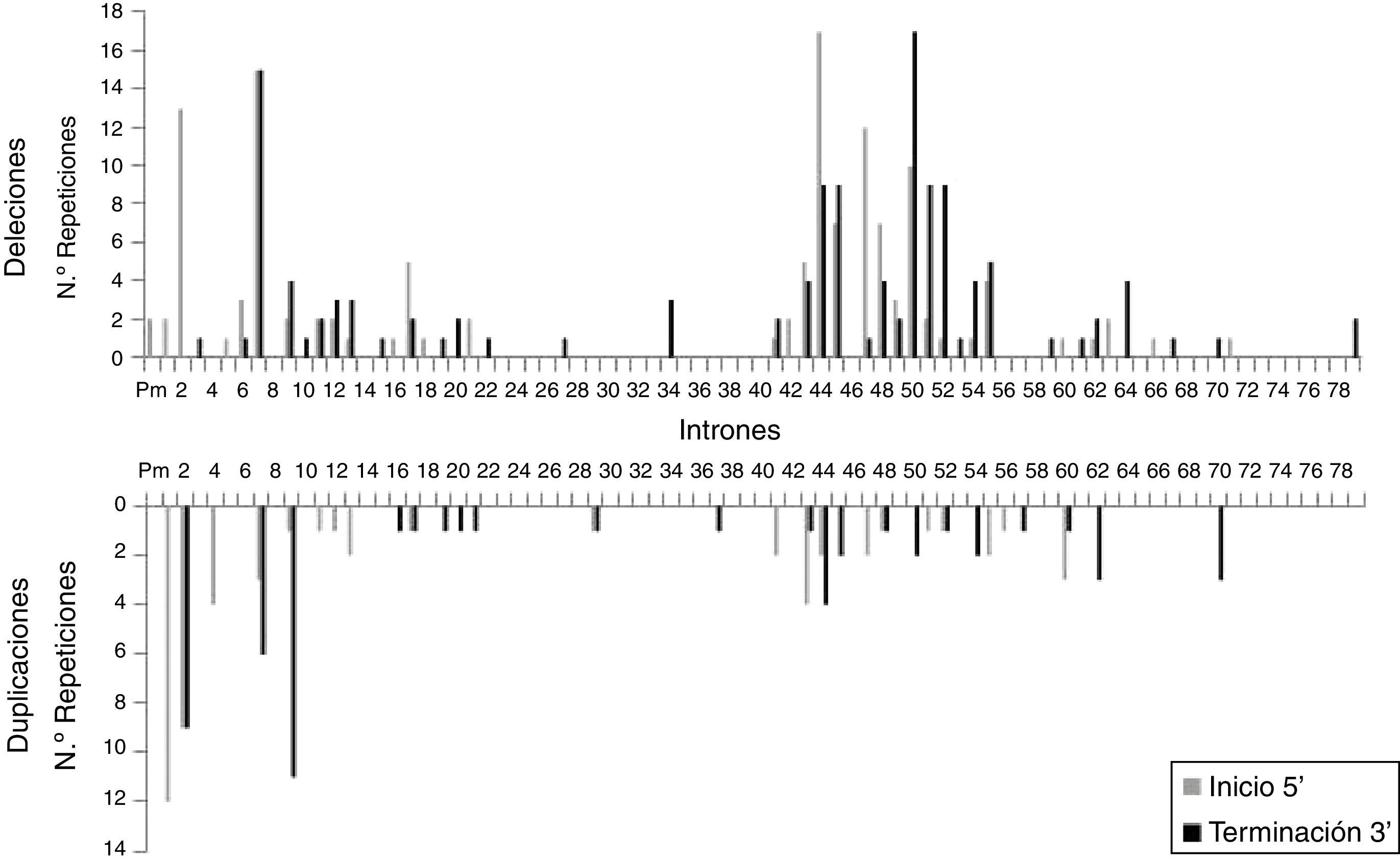
Se analizó la frecuencia con la que las zonas intrónicas de DMD están implicadas en los puntos de corte de las grandes deleciones y duplicaciones, tanto en los inicios 5′ como en las terminaciones 3′. En el caso de las grandes deleciones, se observó que las regiones más implicadas son las correspondientes al intrón 7 (30/131), intrón 44 (26/131) e intrón 50 (29/131), que corresponden al 64,9% de los pacientes con deleciones. En el caso de las grandes duplicaciones, las regiones intrónicas más frecuentemente afectadas son las correspondientes al intrón 1 (12/56), intrón 2 (18/56) e intrón 9 (12/56), que corresponden al 75% de los pacientes con duplicaciones. A diferencia de las grandes deleciones, en las que los puntos de corte parecen estar concentrados en las 2 regiones con más mutaciones, en el caso de las grandes duplicaciones los puntos de corte se encuentran dispersos por todo el gen de la distrofina (fig. 2).
Mutaciones puntuales: nonsense y otras mutaciones
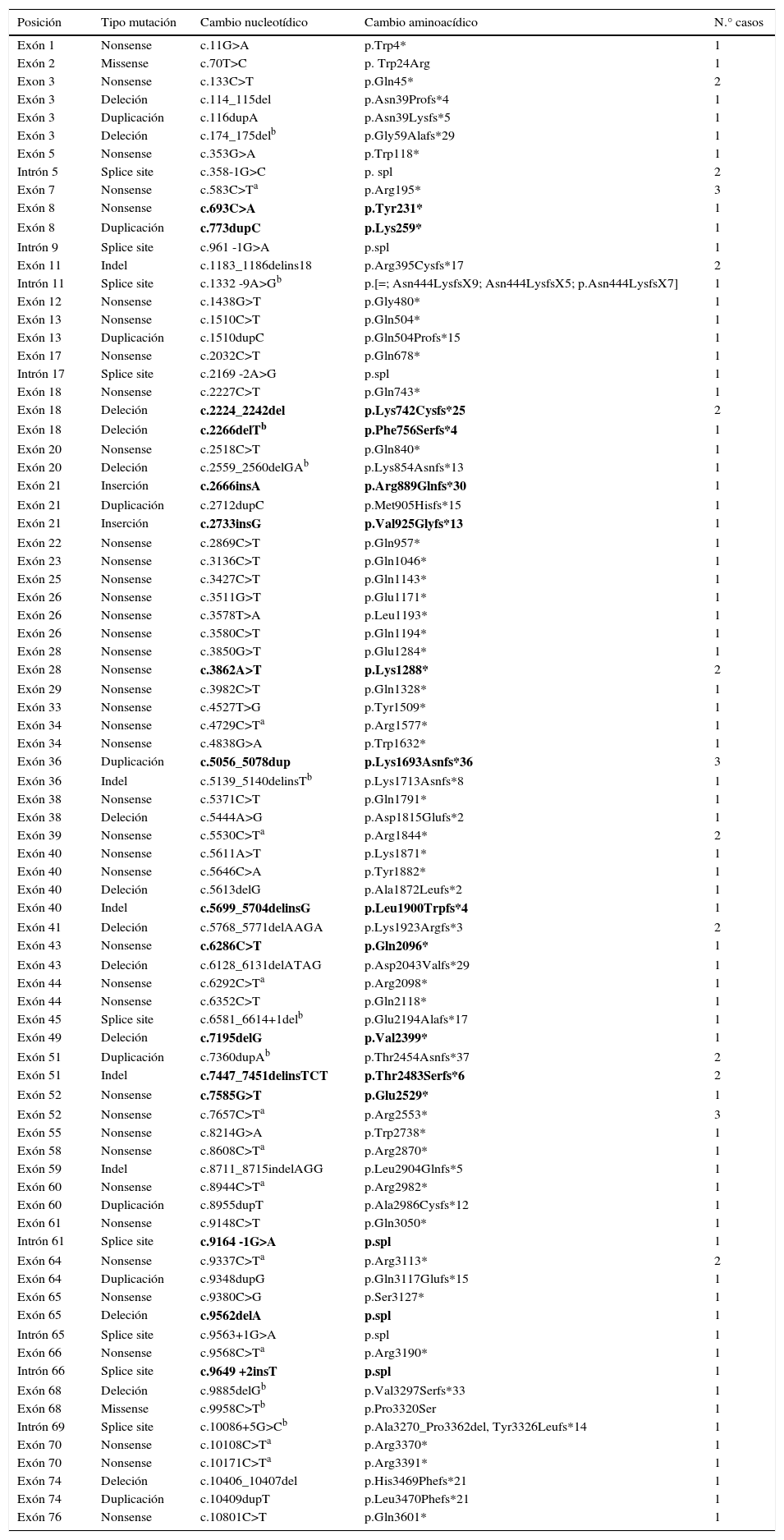
En aquellos pacientes en los que, tras la PCR multiplex o MLPA, no se identificó la mutación en DMD, se realizaron estudios de secuenciación directa por el método Sanger a nivel de ADN, o ARN en caso de disponer de biopsia de músculo. En los casos estudiados más recientemente, el análisis se realizó mediante secuenciación de nueva generación. Se identificaron 97 mutaciones puntuales que corresponden al 34,2% de los 284 pacientes iniciales. Cabe destacar que de estas mutaciones puntuales identificadas, más de la mitad son sustituciones nucleotídicas que generan codones de parada o mutaciones nonsense (49/97, 50,5%) (tabla 1). En este tipo de mutaciones, el cambio de una citosina por timina ocurre en un 67,3% (33/49) de los casos, y en un 51,5% (17/33) de estos casos las citosinas forman parte de dinucleótidos CpG (tabla 2). En 16 casos se analizó la mutación a nivel de transcrito mediante análisis de ARN extraído de la biopsia muscular.
Mutaciones puntuales identificadas en DMD
| Posición | Tipo mutación | Cambio nucleotídico | Cambio aminoacídico | N.° casos |
|---|---|---|---|---|
| Exón 1 | Nonsense | c.11G>A | p.Trp4* | 1 |
| Exón 2 | Missense | c.70T>C | p. Trp24Arg | 1 |
| Exon 3 | Nonsense | c.133C>T | p.Gln45* | 2 |
| Exón 3 | Deleción | c.114_115del | p.Asn39Profs*4 | 1 |
| Exón 3 | Duplicación | c.116dupA | p.Asn39Lysfs*5 | 1 |
| Exón 3 | Deleción | c.174_175delb | p.Gly59Alafs*29 | 1 |
| Exón 5 | Nonsense | c.353G>A | p.Trp118* | 1 |
| Intrón 5 | Splice site | c.358-1G>C | p. spl | 2 |
| Exón 7 | Nonsense | c.583C>Ta | p.Arg195* | 3 |
| Exón 8 | Nonsense | c.693C>A | p.Tyr231* | 1 |
| Exón 8 | Duplicación | c.773dupC | p.Lys259* | 1 |
| Intrón 9 | Splice site | c.961 -1G>A | p.spl | 1 |
| Exón 11 | Indel | c.1183_1186delins18 | p.Arg395Cysfs*17 | 2 |
| Intrón 11 | Splice site | c.1332 -9A>Gb | p.[=; Asn444LysfsX9; Asn444LysfsX5; p.Asn444LysfsX7] | 1 |
| Exón 12 | Nonsense | c.1438G>T | p.Gly480* | 1 |
| Exón 13 | Nonsense | c.1510C>T | p.Gln504* | 1 |
| Exón 13 | Duplicación | c.1510dupC | p.Gln504Profs*15 | 1 |
| Exón 17 | Nonsense | c.2032C>T | p.Gln678* | 1 |
| Intrón 17 | Splice site | c.2169 -2A>G | p.spl | 1 |
| Exón 18 | Nonsense | c.2227C>T | p.Gln743* | 1 |
| Exón 18 | Deleción | c.2224_2242del | p.Lys742Cysfs*25 | 2 |
| Exón 18 | Deleción | c.2266delTb | p.Phe756Serfs*4 | 1 |
| Exón 20 | Nonsense | c.2518C>T | p.Gln840* | 1 |
| Exón 20 | Deleción | c.2559_2560delGAb | p.Lys854Asnfs*13 | 1 |
| Exón 21 | Inserción | c.2666insA | p.Arg889Glnfs*30 | 1 |
| Exón 21 | Duplicación | c.2712dupC | p.Met905Hisfs*15 | 1 |
| Exón 21 | Inserción | c.2733insG | p.Val925Glyfs*13 | 1 |
| Exón 22 | Nonsense | c.2869C>T | p.Gln957* | 1 |
| Exón 23 | Nonsense | c.3136C>T | p.Gln1046* | 1 |
| Exón 25 | Nonsense | c.3427C>T | p.Gln1143* | 1 |
| Exón 26 | Nonsense | c.3511G>T | p.Glu1171* | 1 |
| Exón 26 | Nonsense | c.3578T>A | p.Leu1193* | 1 |
| Exón 26 | Nonsense | c.3580C>T | p.Gln1194* | 1 |
| Exón 28 | Nonsense | c.3850G>T | p.Glu1284* | 1 |
| Exón 28 | Nonsense | c.3862A>T | p.Lys1288* | 2 |
| Exón 29 | Nonsense | c.3982C>T | p.Gln1328* | 1 |
| Exón 33 | Nonsense | c.4527T>G | p.Tyr1509* | 1 |
| Exón 34 | Nonsense | c.4729C>Ta | p.Arg1577* | 1 |
| Exón 34 | Nonsense | c.4838G>A | p.Trp1632* | 1 |
| Exón 36 | Duplicación | c.5056_5078dup | p.Lys1693Asnfs*36 | 3 |
| Exón 36 | Indel | c.5139_5140delinsTb | p.Lys1713Asnfs*8 | 1 |
| Exón 38 | Nonsense | c.5371C>T | p.Gln1791* | 1 |
| Exón 38 | Deleción | c.5444A>G | p.Asp1815Glufs*2 | 1 |
| Exón 39 | Nonsense | c.5530C>Ta | p.Arg1844* | 2 |
| Exón 40 | Nonsense | c.5611A>T | p.Lys1871* | 1 |
| Exón 40 | Nonsense | c.5646C>A | p.Tyr1882* | 1 |
| Exón 40 | Deleción | c.5613delG | p.Ala1872Leufs*2 | 1 |
| Exón 40 | Indel | c.5699_5704delinsG | p.Leu1900Trpfs*4 | 1 |
| Exón 41 | Deleción | c.5768_5771delAAGA | p.Lys1923Argfs*3 | 2 |
| Exón 43 | Nonsense | c.6286C>T | p.Gln2096* | 1 |
| Exón 43 | Deleción | c.6128_6131delATAG | p.Asp2043Valfs*29 | 1 |
| Exón 44 | Nonsense | c.6292C>Ta | p.Arg2098* | 1 |
| Exón 44 | Nonsense | c.6352C>T | p.Gln2118* | 1 |
| Exón 45 | Splice site | c.6581_6614+1delb | p.Glu2194Alafs*17 | 1 |
| Exón 49 | Deleción | c.7195delG | p.Val2399* | 1 |
| Exón 51 | Duplicación | c.7360dupAb | p.Thr2454Asnfs*37 | 2 |
| Exón 51 | Indel | c.7447_7451delinsTCT | p.Thr2483Serfs*6 | 2 |
| Exón 52 | Nonsense | c.7585G>T | p.Glu2529* | 1 |
| Exón 52 | Nonsense | c.7657C>Ta | p.Arg2553* | 3 |
| Exón 55 | Nonsense | c.8214G>A | p.Trp2738* | 1 |
| Exón 58 | Nonsense | c.8608C>Ta | p.Arg2870* | 1 |
| Exón 59 | Indel | c.8711_8715indelAGG | p.Leu2904Glnfs*5 | 1 |
| Exón 60 | Nonsense | c.8944C>Ta | p.Arg2982* | 1 |
| Exón 60 | Duplicación | c.8955dupT | p.Ala2986Cysfs*12 | 1 |
| Exón 61 | Nonsense | c.9148C>T | p.Gln3050* | 1 |
| Intrón 61 | Splice site | c.9164 -1G>A | p.spl | 1 |
| Exón 64 | Nonsense | c.9337C>Ta | p.Arg3113* | 2 |
| Exón 64 | Duplicación | c.9348dupG | p.Gln3117Glufs*15 | 1 |
| Exón 65 | Nonsense | c.9380C>G | p.Ser3127* | 1 |
| Exón 65 | Deleción | c.9562delA | p.spl | 1 |
| Intrón 65 | Splice site | c.9563+1G>A | p.spl | 1 |
| Exón 66 | Nonsense | c.9568C>Ta | p.Arg3190* | 1 |
| Intrón 66 | Splice site | c.9649 +2insT | p.spl | 1 |
| Exón 68 | Deleción | c.9885delGb | p.Val3297Serfs*33 | 1 |
| Exón 68 | Missense | c.9958C>Tb | p.Pro3320Ser | 1 |
| Intrón 69 | Splice site | c.10086+5G>Cb | p.Ala3270_Pro3362del, Tyr3326Leufs*14 | 1 |
| Exón 70 | Nonsense | c.10108C>Ta | p.Arg3370* | 1 |
| Exón 70 | Nonsense | c.10171C>Ta | p.Arg3391* | 1 |
| Exón 74 | Deleción | c.10406_10407del | p.His3469Phefs*21 | 1 |
| Exón 74 | Duplicación | c.10409dupT | p.Leu3470Phefs*21 | 1 |
| Exón 76 | Nonsense | c.10801C>T | p.Gln3601* | 1 |
En negrita, las mutaciones puntuales no descritas.
El resto de las mutaciones identificadas representa el 16,9% del total de pacientes e incluye 15 pequeñas deleciones, 12 pequeñas duplicaciones, 10 alteraciones de splicing, 7 indels, 2 pequeñas inserciones y 2 sustituciones missense. El rango de tamaño de estas variaciones oscila entre la sustitución de un único nucleótido hasta la deleción de 23 nucleótidos. En 10 casos se confirmó la mutación a nivel de transcrito (tabla 2).
Nuevas mutaciones en el gen DMDEn el estudio realizado se identificaron 23 alteraciones genéticas en el gen DMD no descritas con anterioridad: 7 corresponden a grandes deleciones (deleción de los exones 2 al 15; 3 al 45; 56 al 61; 56 al 64; 63 al 64; 64 al 70 y deleción del exón 67) y 16 a mutaciones puntuales (4 sustituciones nonsense, 4 pequeñas deleciones, 2 pequeñas duplicaciones, 2 indels, 2 pequeñas inserciones y 2 variaciones de splicing) (tabla 2).
Distribución geográfica de los casos estudiadosLa serie de casos recogida se sitúa en torno al 26% (284/1091) de los casos de DMD que se esperaría tener en España en el periodo estudiado, teniendo en cuenta la prevalencia global estimada para DMD de 4,78 (IC del 95%, 1,94-11,81) casos por cada 100.000 varones24 y los datos poblacionales facilitados por el Instituto Nacional de Estadística (22.820.775 varones en enero de 2015). La distribución geográfica de los casos observados en este estudio es heterogénea, siendo las comunidades autónomas con más casos de DMD diagnosticados genéticamente las correspondientes a los centros de referencia que participan en el estudio: Cataluña, Comunidad de Madrid, Andalucía, Comunidad Valenciana, País Vasco, Galicia y Cantabria.
DiscusiónLa DMD es una de las enfermedades neuromusculares más frecuentes, con patrón de herencia ligada al cromosoma X. Es la variante fenotípica más grave asociada al gen de la distrofina y se suele diagnosticar entre los 2 y 5 años de edad. La ausencia de la distrofina da lugar a la degeneración progresiva de la fibra muscular con fibrosis y sustitución adiposa terminal, lo que determina una debilidad muscular progresiva y pérdida de la deambulación alrededor de los 13 años de edad. A diferencia de otras distrofinopatías, el cuadro clínico de los pacientes afectados de DMD es relativamente uniforme, y las diferencias en la evolución individual se deben especialmente a la afectación cardíaca asociada y a las terapias o cuidados particulares otorgados a los pacientes a distintos niveles: rehabilitación, seguimiento cardíaco, respiratorio, ortopédico, psicosocial y nutricional14,29,30.
En este artículo se han recogido los casos diagnosticados de DMD en los principales laboratorios españoles de genética molecular, tratando de profundizar en las características genéticas que presentan. El análisis mutacional de los 284 pacientes estudiados confirma la gran heterogeneidad genética de la DMD, identificándose 179 mutaciones distintas en DMD que se pueden clasificar en 3 categorías: grandes deleciones (con 71 tipos distintos en 131 pacientes); grandes duplicaciones (con 27 tipos distintos en 56 pacientes), y mutaciones puntuales (con 81 tipos distintos en 97 pacientes).
De la serie de afectados de DMD estudiados, un 46,1% de los casos presentó grandes deleciones. Este porcentaje es menor de lo publicado en estudios previos31. Esto podría deberse a un sesgo en la selección de los pacientes, ya que algunos de ellos corresponden a portadores de mutaciones puntuales que no fueron detectadas antes de 2007, y cuyo análisis se retomó posteriormente empleando nuevas tecnologías11. Las deleciones más frecuentes en la serie analizada fueron las de los exones 51 (7,6%) y 45 (5,3%), y se observó una agrupación de las deleciones en los 2 puntos calientes del gen: el 38,2% en la región proximal y el 55,9% en la distal. Estos datos están en consonancia con estudios previos publicados sobre grandes series de pacientes con DMD11,12,32,33.
En cuanto a las duplicaciones exónicas, representan un 19,7% de los pacientes estudiados, y un 17,9% de ellos portan la duplicación del exón 2. Estos datos son similares a los reportados en estudios anteriores12,34. Analizando la distribución de las grandes duplicaciones identificadas, también tienden a la agrupación en las regiones calientes del gen de la distrofina, aunque, en este caso, parecen tener una mayor concentración en la región proximal que en la distal (el 67,9 y el 28,6%, respectivamente). Estos datos son concordantes con estudios previos11,12,32,33.
El patrón de distribución de los puntos de corte de las mutaciones varía entre grandes deleciones y duplicaciones. En las deleciones los puntos de corte se concentran en las regiones calientes del gen de la distrofina, especialmente en la región distal, y, en cambio, los puntos de corte implicados en las duplicaciones se localizan principalmente en la región proximal, estando el resto más dispersos a lo largo del gen. Por otra parte, aunque los puntos de corte en las deleciones coinciden fundamentalmente con los intrones de gran tamaño, la longitud de la región intrónica no debe considerarse como el único determinante en el perfil de punto de corte de deleciones en el gen DMD35. En nuestra serie, aunque el intrón 7, de unos 110kb, es el que presenta un mayor número de puntos de corte, el segundo con una elevada concentración de puntos de corte es el intrón 50, de mucho menor tamaño (46kb).
A diferencia de las grandes deleciones y duplicaciones, las mutaciones puntuales, que representan en su conjunto el 34,2% de los casos estudiados, se distribuyen uniformemente a lo largo del gen DMD y son muy poco recurrentes, siendo la mayoría de ellas identificadas en un único paciente. Dentro de las mutaciones puntuales, las sustituciones sin sentido o nonsense son las más frecuentes, con un 50,5% de los casos estudiados. Un dato a resaltar es el elevado porcentaje de sustituciones de citosina por timina, que corresponden a citosinas implicadas en los dinucleótidos CpG (51,5%). Además, en la distrofina existen 29 codones CGA (arginina), y en este estudio se ha identificado la transición de la citosina a timina en 11 de ellos. Varios estudios publicados con anterioridad ya observaron esta tendencia a la transición a timina de las citosinas metiladas de los dinucleótidos CpG por desaminación espontánea, por lo que estos dinucleótidos CpG son puntos calientes de mutaciones en el gen de la distrofina11,32,33,36,37.
La hipótesis de la pauta de lectura establecida por Monaco et al.8 postula que mutaciones que alteran la pauta de lectura dan lugar a la ausencia de distrofina generando el fenotipo de DMD. En esta serie, 254 pacientes son portadores de mutaciones que rompen la pauta de lectura (89,4%) y 30 pacientes presentan mutaciones que mantienen la pauta de lectura (10,6%), siendo estas últimas excepciones a la regla. En el caso de las grandes deleciones, se ajustan a la hipótesis el 88,5% de los casos y no se ajustan el 11,5% de ellos. Una posible explicación a esta excepción sería que el punto de corte de la deleción se produjese dentro de regiones exónicas, lo que sí podría dar lugar a una alteración de la pauta de lectura38. Por otra parte, algunas deleciones que mantienen la pauta de lectura afectan a dominios de unión de la distrofina (dominio de unión a la actina y dominio rico en cisteína), lo que afectaría seriamente al correcto funcionamiento de la proteína33. Hay que considerar que la aplicación de esta hipótesis sobre las duplicaciones no es totalmente fiable, ya que se presupone una duplicación en tándem, lo que no siempre ocurre, siendo necesario un estudio del mARN para comprobar esta afirmación34. Otras mutaciones que escapan a la regla de Monaco son las de tipo missense, que, en principio, se asociarían con un fenotipo menos grave de distrofinopatía. En cambio, las 2 mutaciones de este tipo identificadas en nuestra serie de pacientes se localizan en los dominios de unión de la distrofina: la mutación p. Trp24Arg se localiza en el dominio ABD y la mutación p. Pro3320Ser en el dominio CRD. En cuanto a las alteraciones de splicing, estudios previos en mARN han demostrado que este tipo de variaciones suele provocar alteraciones de la pauta de lectura11.
Sobre la base de los resultados obtenidos, creemos que el algoritmo de diagnóstico aplicado en los centros que participaron en este estudio es el más adecuado para el diagnóstico molecular de la DMD, permitiendo genotipificar, mediante MLPA y/o PCR multiplex, el 65,8% de los casos, aplicando, en segundo término, tecnologías de secuenciación para completar el análisis de mutaciones puntuales en DMD.
En los últimos años se ha desarrollado un elevado número de ensayos clínicos dirigidos al tratamiento de los pacientes de DMD, que incluyen estrategias de sustitución génica, corrección del gen DMD o modificación del producto génico, y que pueden ser aplicados únicamente en pacientes con determinados tipos de mutaciones39,40. Una de las terapias en estudio más prometedora en la actualidad se basa en la relectura de mutaciones sin sentido (nonsense read-through) que trata de suprimir el efecto de una sustitución que genera un codón de parada mediante el tratamiento con PTC12441,42. Un 17,2% (110/284) de los casos de DMD de la serie son portadores de mutaciones sin sentido, por lo que serían potencialmente beneficiarios de este tipo de tratamiento. Otra terapia destacada en el tratamiento de pacientes de DMD consiste en el salto de exón (exon skipping), de forma que se restauraría la pauta de lectura mediante la supresión de uno o 2 exones. El mecanismo de salto de exón 51 es el más desarrollado, seguido de los exones 44, 45, 52, 53 y 5543,44. Tras el análisis genético, se encontró que un 21,5% (61/284) de los pacientes presentan deleciones que, mediante omisión de alguno de estos exones, podrían restablecer la pauta de lectura de la distrofina. Esto hace que el 38,7% (110/284) de los pacientes de nuestra serie sean a priori susceptibles de participar en alguno de los recientes ensayos clínicos a expensas de los criterios de inclusión/exclusión. Con esto queremos resaltar la gran importancia de identificar el tipo de mutación que presentan los pacientes con DMD con el fin de seleccionar los que podrían eventualmente ser incluidos en un ensayo clínico y en qué tipo de ensayo. Por otra parte, el diagnóstico genético es igualmente importante para la prevención de nuevos casos en una familia afectada.
Otro de los objetivos de este artículo fue analizar la distribución geográfica de afectos de DMD en España en el periodo 2007-2014. Observamos que solo hemos logrado reunir el 26% de los casos que se esperaría obtener en la población española, lo que pone de manifiesto, al margen de que existan posibles casos no identificados, el infradiagnóstico genético de DMD en España. Esto puede ser debido, en parte, a las dificultades de acceso a los centros de referencia y posiblemente también a una insuficiente relación entre clínicos y genetistas. Una mayor coordinación entre ellos ayudaría a agilizar y mejorar el diagnóstico genético en las distrofinopatías y otras enfermedades neuromusculares, profundizando en la correlación clínico-patológica y genética y favoreciendo la aplicación de posibles tratamientos en estos pacientes con enfermedades tan graves y que, hasta el momento, cuentan con tan escasas ayudas terapéuticas.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Los autores agradecen a la Dra. Susana Teijeira (Complexo Hospitalario Universitario de Vigo e Instituto de Investigación Biomédica Ourense-Pontevedra-Vigo, IBI) y a la Dra. Paloma Martínez Montero (Instituto de Genética Médica y Molecular (INGEMM), Hospital Universitario La Paz, Madrid) su colaboración en la elaboración de este trabajo, así como a todos los centros que participaron en el mismo.

