





La enfermedad de Steinert o distrofia miotónica tipo 1 (DM1), (OMIM 160900) es la miopatía más prevalente en el adulto. Es una enfermedad multisistémica con alteración de prácticamente todos los órganos y tejidos y una variabilidad fenotípica muy amplia, lo que implica que deba ser atendida por diferentes especialistas que dominen las alteraciones más importantes. En los últimos años se ha avanzado de manera exponencial en el conocimiento de la enfermedad y en su manejo. El objetivo de la guía es establecer recomendaciones para el diagnóstico, el pronóstico, el seguimiento y el tratamiento de las diferentes alteraciones de la DM1.
Material y métodosEsta guía de consenso se ha realizado de manera multidisciplinar. Se ha contado con neurólogos, neumólogos, cardiólogos, endocrinólogos, neuropediatras y genetistas que han realizado una revisión sistemática de la literatura.
RecomendacionesSe recomienda realizar un diagnóstico genético con cuantificación precisa de tripletes CTG. Los pacientes con DM1 deben seguir control cardiológico y neumológico de por vida. Antes de cualquier cirugía con anestesia general debe realizarse una evaluación respiratoria. Debe monitorizarse la presencia de síntomas de disfagia periódicamente. Debe ofrecerse consejo genético a los pacientes con DM1 y a sus familiares.
ConclusiónLa DM1 es una enfermedad multisistémica que requiere un seguimiento en unidades especializadas multidisciplinares.
Steinert's disease or myotonic dystrophy type 1 (MD1), (OMIM 160900), is the most prevalent myopathy in adults. It is a multisystemic disorder with dysfunction of virtually all organs and tissues and a great phenotypical variability, which implies that it has to be addressed by different specialities with experience in the disease. The knowledge of the disease and its management has changed dramatically in recent years. This guide tries to establish recommendations for the diagnosis, prognosis, follow-up and treatment of the complications of MD1.
Material and methodsConsensus guide developed through a multidisciplinary approach with a systematic literature review. Neurologists, pulmonologists, cardiologists, endocrinologists, neuropaediatricians and geneticists have participated in the guide.
RecommendationsThe genetic diagnosis should quantify the number of CTG repetitions. MD1 patients need cardiac and respiratory lifetime follow-up. Before any surgery under general anaesthesia, a respiratory evaluation must be done. Dysphagia must be screened periodically. Genetic counselling must be offered to patients and relatives.
ConclusionMD1 is a multisystemic disease that requires specialised multidisciplinary follow-up.
La distrofia miotónica tipo 1, enfermedad de Steinert, enfermedad de Steinert-Curschmann, enfermedad de Batten-Gibb, miotonía atrófica o DM1 (OMIM 160900, ORPHA 273, CIE-9-MC 359.21, CIE-10 G71.1, CIE-11 8C71.0) es la miopatía más prevalente en el adulto1,2. Es una enfermedad autosómica dominante producida por la expansión de tripletes CTG en la región no codificante del gen DMPK (proteincinasa de la distrofia miotónica), localizada en el brazo largo del cromosoma 19 (19q13.3). Es una enfermedad tradicionalmente diagnosticada por los neurólogos por sus características alteraciones neuromusculares, pero que produce también alteraciones sistémicas. Ha llegado a ser considerada una de las enfermedades con mayor variedad fenotípica que existen. Esta variabilidad clínica en las manifestaciones, tanto en cualidad como en cantidad, conlleva una necesidad de tratamiento personalizado de cada paciente, así como de un conocimiento adecuado y profundo de las alteraciones de cada órgano y sistema para poder ofrecer a los pacientes el seguimiento y tratamiento más apropiados.
MetodologíaEl objetivo de esta guía es servir de referencia a los profesionales implicados en el diagnóstico y seguimiento de los pacientes de DM1. En ella se hacen recomendaciones específicas en cuanto a diagnóstico, el seguimiento y el tratamiento de las alteraciones de la enfermedad.
El grupo que ha desarrollado esta guía incluye a especialistas de todos los grupos profesionales implicados en la atención de los pacientes con DM1: neurólogos, neumólogos, cardiólogos, endocrinólogos, neuropediatras y genetistas. Se han tenido en cuenta la visión del paciente y sus familiares, sus preferencias, y los usuarios de la guía están claramente definidos.
Para realizar esta guía se llevó a cabo una búsqueda bibliográfica en las bases de datos Biblioteca Cochrane, Cochrane Plus, Embase, Pubmed-Medline y ECA LOST siguiendo el método PICO (pacientes-intervención-comparación-resultados) que recomienda el grupo de trabajo de guías de práctica clínica del Sistema Nacional de Salud3. Se utilizaron los términos de búsqueda «myotonic dystrophy», «myotonic dystrophy type 1» y «Steinert's disease», sin especificar fecha. También se utilizaron las referencias bibliográficas de la literatura encontrada. Se seleccionaron los artículos más recientes y de mayor calidad científica en español, inglés y francés.
Partiendo de los artículos revisados, la guía fue elaborada en subgrupos de trabajo que se encargaron de cada uno de los temas correspondientes: diagnóstico, alteraciones motoras y cognitivas, alteraciones cardíacas, alteraciones respiratorias, otras alteraciones y DM1 en el niño. Cada subgrupo elaboró un documento inicial que fue discutido y consensuado en la reunión ad hoc celebrada en el seno de la Reunión de Primavera del Grupo de Estudio de Enfermedades Neuromusculares, en Santiago de Compostela, el 29 de abril de 2017. Posteriormente se mantuvieron diversas conversaciones para dar una forma definitiva a la guía.
Los autores representan al comité de expertos nacional sobre la DM1, que es un panel de médicos de diferentes especialidades con amplia experiencia clínica en la enfermedad. Los autores son responsables de la interpretación presentada en esta revisión. Todos los autores han aprobado la versión final de este manuscrito y se responsabilizan totalmente de su contenido.
Se ha utilizado la herramienta AGREE II4 para evaluar y garantizar la calidad, la claridad, el rigor, la aplicabilidad y la independencia editorial de esta guía.
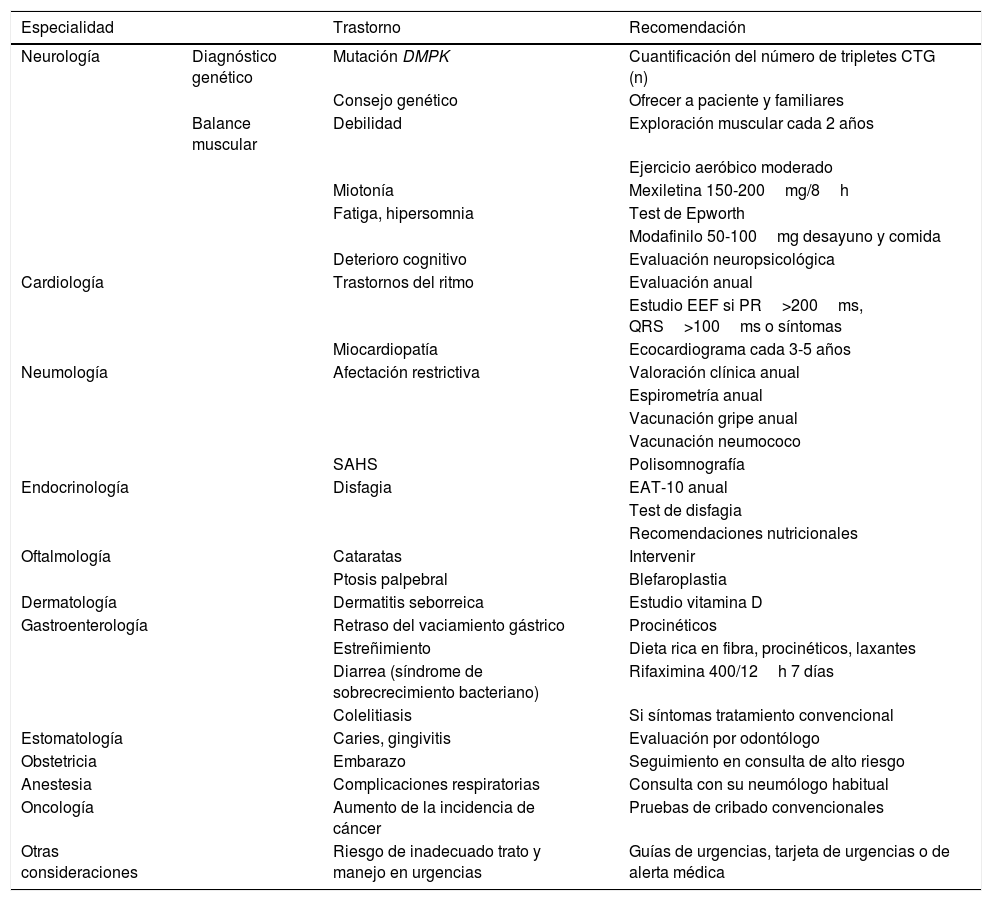
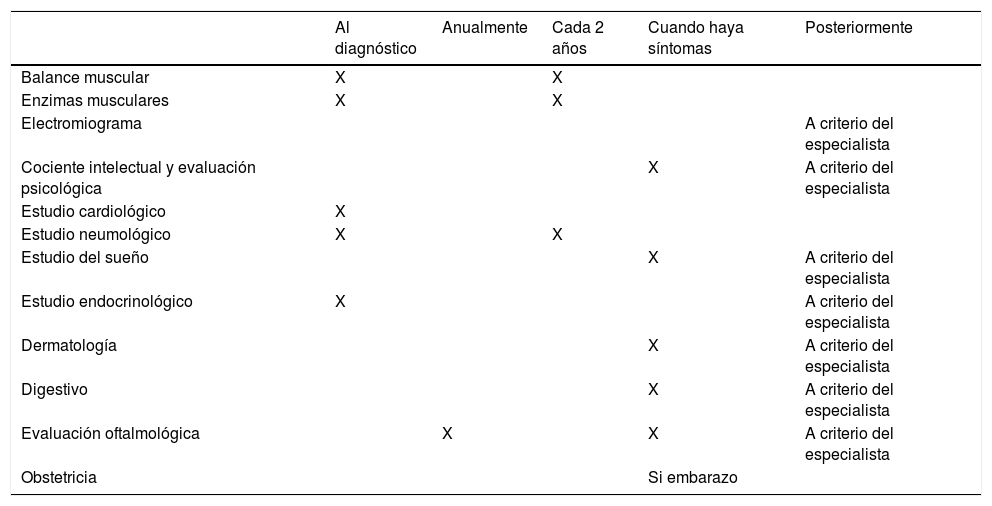
Las recomendaciones se resumen en la tabla 1 y una propuesta de la periodicidad de las evaluaciones en la tabla 2.
Resumen de recomendaciones
| Especialidad | Trastorno | Recomendación | |
|---|---|---|---|
| Neurología | Diagnóstico genético | Mutación DMPK | Cuantificación del número de tripletes CTG (n) |
| Consejo genético | Ofrecer a paciente y familiares | ||
| Balance muscular | Debilidad | Exploración muscular cada 2 años | |
| Ejercicio aeróbico moderado | |||
| Miotonía | Mexiletina 150-200mg/8h | ||
| Fatiga, hipersomnia | Test de Epworth | ||
| Modafinilo 50-100mg desayuno y comida | |||
| Deterioro cognitivo | Evaluación neuropsicológica | ||
| Cardiología | Trastornos del ritmo | Evaluación anual | |
| Estudio EEF si PR>200ms, QRS>100ms o síntomas | |||
| Miocardiopatía | Ecocardiograma cada 3-5 años | ||
| Neumología | Afectación restrictiva | Valoración clínica anual | |
| Espirometría anual | |||
| Vacunación gripe anual | |||
| Vacunación neumococo | |||
| SAHS | Polisomnografía | ||
| Endocrinología | Disfagia | EAT-10 anual | |
| Test de disfagia | |||
| Recomendaciones nutricionales | |||
| Oftalmología | Cataratas | Intervenir | |
| Ptosis palpebral | Blefaroplastia | ||
| Dermatología | Dermatitis seborreica | Estudio vitamina D | |
| Gastroenterología | Retraso del vaciamiento gástrico | Procinéticos | |
| Estreñimiento | Dieta rica en fibra, procinéticos, laxantes | ||
| Diarrea (síndrome de sobrecrecimiento bacteriano) | Rifaximina 400/12h 7 días | ||
| Colelitiasis | Si síntomas tratamiento convencional | ||
| Estomatología | Caries, gingivitis | Evaluación por odontólogo | |
| Obstetricia | Embarazo | Seguimiento en consulta de alto riesgo | |
| Anestesia | Complicaciones respiratorias | Consulta con su neumólogo habitual | |
| Oncología | Aumento de la incidencia de cáncer | Pruebas de cribado convencionales | |
| Otras consideraciones | Riesgo de inadecuado trato y manejo en urgencias | Guías de urgencias, tarjeta de urgencias o de alerta médica |
Evaluaciones periódicas recomendadas
| Al diagnóstico | Anualmente | Cada 2 años | Cuando haya síntomas | Posteriormente | |
|---|---|---|---|---|---|
| Balance muscular | X | X | |||
| Enzimas musculares | X | X | |||
| Electromiograma | A criterio del especialista | ||||
| Cociente intelectual y evaluación psicológica | X | A criterio del especialista | |||
| Estudio cardiológico | X | ||||
| Estudio neumológico | X | X | |||
| Estudio del sueño | X | A criterio del especialista | |||
| Estudio endocrinológico | X | A criterio del especialista | |||
| Dermatología | X | A criterio del especialista | |||
| Digestivo | X | A criterio del especialista | |||
| Evaluación oftalmológica | X | X | A criterio del especialista | ||
| Obstetricia | Si embarazo |
Los pacientes con formas típicas de la enfermedad pueden presentar una ligera elevación de CK. En los individuos asintomáticos, los niveles de CK suelen ser normales.
Estudio neurofisiológicoEl estudio electromiográfico detecta descargas miotónicas típicas. Estas consisten en salvas de ondas positivas o fibrilaciones, con frecuencia y amplitud decreciente, que les da un aspecto y un sonido característico, como de un avión cayendo en picado. El patrón electromiográfico muestra rasgos miopáticos, con potenciales polifásicos de pequeña amplitud y un patrón interferencial precoz. Aunque estas alteraciones se pueden observar en cualquier músculo, son más evidentes en la musculatura distal.
En el estudio electroneurográfico se aprecia una reducción en la amplitud del potencial evocado motor compuesto.
El test de ejercicio corto muestra una caída precoz, inmediatamente tras el esfuerzo, en la amplitud del potencial evocado motor compuesto, similar a lo que ocurre en las canalopatías por alteración en el gen del canal de cloro (miotonías de Thomsen y Becker). Esto no es de extrañar, dado que se asume que la miotonía de la DM1 se produce por interferencia con la transcripción del gen del canal de cloro (CLCN-1), mutado en las miotonías congénitas. Este hallazgo ayuda a diferenciar la DM1 de la DM2 (distrofia miotónica proximal o PROMM), en la que no se observa modificación en la amplitud de potencial evocado motor compuesto con el esfuerzo.
Biopsia muscularLa biopsia muscular no ha sido nunca un procedimiento clave para el diagnóstico de DM1. No hay datos histopatológicos patognomónicos. No obstante, la asociación de gran cantidad de núcleos centrales, aglomeraciones de núcleos picnóticos, masas sarcoplásmicas, existencia de fibras en anillo y apolilladas, y atrofia selectiva de fibras tipo 1 es muy sugestiva de distrofia miotónica de Steinert. Estos hallazgos son similares a los encontrados en la DM2, salvo que en esta predomina la atrofia de fibras tipo 2, al contrario de lo que ocurre en la DM1.
En cualquier caso, dada la accesibilidad y especificidad del diagnóstico genético, no es necesario realizar una biopsia muscular a un paciente con sospecha clínica de DM1.
Resonancia magnética nuclear muscularA pesar de que la resonancia magnética nuclear (RMN) muscular se ha implantado como un procedimiento diagnóstico fundamental en el estudio de las enfermedades neuromusculares, en la DM1, dada la expresividad de los rasgos clínicos y la accesibilidad del diagnóstico genético, existe poca experiencia con esta técnica. En miembros superiores se ha publicado una afectación de los músculos flexor profundo de los dedos, flexor superficial de los dedos, flexor largo del pulgar, extensores del pulgar, abductor corto del pulgar y, luego, de la cabeza lateral del tríceps braquial y el infraespinoso. En miembros inferiores se observa una afectación inicial en tibial anterior y, luego, de semimembranoso, vasto intermedio y gemelo medial. También se puede detectar una afectación precoz de la musculatura paravertebral. Existe una buena correlación entre la expresión clínica y las alteraciones encontradas en la RMN muscular.
Diagnóstico genéticoLa confirmación diagnóstica de la DM1 pasa por la detección de la alteración genética que produce la enfermedad. Esta consiste en un aumento del número de repeticiones de un triplete CTG situado en una región no codificante del extremo 3’ del gen DMPK, localizado en el brazo largo del cromosoma 19 (19q13.3).
Los individuos normales tienen entre 5 y 34 repeticiones del triplete CTG. Las personas que tienen entre 35 y 49 repeticiones (premutados) no presentan síntomas de la enfermedad, pero pueden transmitir un mayor número de repeticiones a su descendencia, que puede alcanzar ya el rango de mutación. Por encima de 50 repeticiones, se considera que el alelo está mutado y la persona que lo porta sufre la enfermedad, con una gravedad que se correlaciona con el número de tripletes. En general, cuanto mayor es el número de repeticiones CTG la enfermedad se inicia más precozmente y se manifiesta con más síntomas y más intensos. La correlación genotipo-fenotipo es mayor por debajo de 400 repeticiones. Por encima de este número, la inestabilidad en la transmisión mitótica provoca errores en la transmisión del fragmento de una célula a sus hijas, de modo que cada individuo es un mosaico, con expansiones diferentes en cada tejido. Por este motivo, el número de tripletes medido en linfocitos puede no ser proporcional al que haya en otros tejidos, como el músculo. De hecho, el número de CTG en músculo esquelético suele ser entre 2 y 13 veces el encontrado en leucocitos.
Habitualmente, los pacientes con un número de repeticiones entre 50 y 100 CTG tienen formas leves de la enfermedad, con cataratas a los 50-60 años y/o leve miotonía. Los pacientes con la forma clásica de la enfermedad suelen tener entre 100 y 1.000 CTG, con un fenotipo más grave cuanto mayor es la expansión. Finalmente, la mayoría de las distrofias miotónicas congénitas tiene expansiones por encima de 1.000 CTG. No obstante, la correlación no es perfecta y pueden verse fenotipos más graves o más leves de los que corresponderían al rango de repeticiones CTG indicado. Además, el tamaño de la expansión puede cambiar con la edad, por lo que la correlación genotipo-fenotipo más aproximada se observa cuando la evaluación clínica es cercana en el tiempo a la determinación del número de repeticiones CTG.
Detección de la expansión CTGEl estudio de la expansión del fragmento CTG se hace a partir de una muestra de 10ml de sangre en EDTA. La determinación del tamaño de este fragmento en otros tejidos como el músculo no aporta nada al diagnóstico.
Mediante polymerase chain reaction (PCR, «reacción en cadena de la polimerasa») se detectan expansiones de hasta 180 repeticiones CTG. La Triple-repeat Primed-PCR (TP-PCR) es una técnica barata que detecta si el número de repeticiones CTG está en el rango normal o patológico, pero no permite cuantificarlas. Para determinar el número de repeticiones por encima de 180 CTG es necesario emplear técnicas de Southern Blot.
El estudio molecular detecta prácticamente el 100% de las variantes patógenas, de modo que la sensibilidad y especificidad del estudio genético son cercanas al 100%. No es suficiente indicar si la expansión del fragmento es mayor o menor de 50 CTG. Es fundamental cuantificar el número de repeticiones CTG dado el valor pronóstico de esta determinación.
Consejo genético. AnticipaciónLa DM1 se transmite con un patrón de herencia autosómico dominante. Por tanto, el riesgo que tiene cada hijo de un paciente de heredar la mutación es de un 50%. La penetrancia es muy alta, cercana al 100% a los 50 años de edad, cuando se consideran todas las manifestaciones clínicas de la enfermedad.
La gravedad del fenotipo del hijo dependerá del tamaño del fragmento CTGn que herede. Al ser inestable la transmisión de este fragmento, con tendencia a incrementar el número de tripletes CTG que pasan a la descendencia, los hijos que hereden la mutación suelen presentar formas más graves que sus padres, fenómeno conocido como anticipación clínica. Las expansiones entre 50 y 80 CTG pueden transmitirse durante varias generaciones sin grandes cambios. No obstante, los alelos de este tamaño presentan una mayor inestabilidad cuando el progenitor es un varón. Fuera de este rango, la transmisión por vía materna suele asociarse a un mayor salto intergeneracional, y el incremento en el número de repeticiones CTG puede ser tan masivo que alcance el rango de DM1 congénita. El riesgo de tener un hijo con DM congénita es mayor cuanto mayor es la expansión materna, especialmente por encima de 300 CTG, pero esta posibilidad existe siempre que una mujer pasa la mutación a su descendencia, aun cuando su expansión no sea muy grande. Incluso una mujer asintomática o con una forma mínima de la enfermedad puede tener un hijo con la variante congénita.
Más raro es que se produzcan contracciones en la longitud del fragmento CTG que se transmite. Este fenómeno se da con más frecuencia cuando el transmisor es un varón con fragmentos grandes, por encima de 500 CTG.
Siempre que se diagnostique a un paciente de DM1, debe explicarse el riesgo que tienen sus familiares de sufrir la enfermedad, aun cuando por el momento no presenten ningún síntoma, y proponer un estudio genético para todos los mayores de 18 años. El diagnóstico presintomático en menores de esta edad se considera inapropiado, dado que no existe tratamiento para la enfermedad, niega la autonomía del individuo para decidir lo que quiere o no saber y supone un riesgo de estigmatización y discriminación, así como de generar ansiedad en el niño y sobreprotección por parte de los padres. La necesidad de realizar un adecuado consejo genético es especialmente imperiosa en los casos de mujeres en edad fértil en riesgo, por la posibilidad de tener hijos con formas congénitas de la enfermedad. De todas formas, la detección precoz de todos los portadores de la mutación tiene gran transcendencia, dado el riesgo de sufrir complicaciones graves por la enfermedad, especialmente en la esfera cardiológica. Previamente a la realización del test es recomendable entrevistarse con el individuo en riesgo y explicarle las características de la enfermedad y de su transmisión.
Planificación familiar. Diagnóstico prenatal y preimplantacionalExiste la posibilidad de realizar el diagnóstico en embrión o incluso seleccionar in vitro embriones sanos para su implantación posterior. Estas opciones se deben analizar y ofrecer antes de la gestación.
El diagnóstico prenatal consiste en la realización del estudio genético a partir de una muestra de vellosidad corial extraída entre las semanas 9-12 de gestación. También se puede llevar a cabo mediante amniocentesis en la semana 16. Estos procedimientos tienen un riesgo de un 3-4% de provocar un aborto.
El diagnóstico preimplantacional es una opción que podría evitar este riesgo de aborto, lo que puede tener gran relevancia en una enfermedad en la que la fertilidad está disminuida y se asocia ya a un riesgo de aborto espontáneo elevado. Además, obvia de alguna manera los problemas éticos que para algunas personas puede suponer un aborto terapéutico. No obstante, es un procedimiento que exige un gran esfuerzo a la mujer por lo que supone la estimulación ovárica y la extracción de óvulos. Es recomendable iniciar este procedimiento antes de que la reserva ovárica caiga por causas naturales (en torno a los 38 años) o por la propia enfermedad. Por otro lado, la tasa de embarazos conseguida es relativamente baja y, a menudo, es necesario repetir el proceso varias veces para conseguir culminar con el nacimiento de un niño. Además, con frecuencia se exige la confirmación de que el embrión no es portador de la mutación mediante estudio de vellosidad corial, con lo que el riesgo de aborto asociado al procedimiento sería el mismo que el del diagnóstico prenatal.
Protocolo diagnósticoAnte un paciente con sospecha clínica de DM1, el procedimiento diagnóstico de elección es el estudio genético. El resto de las técnicas mencionadas pueden tener interés desde el punto de vista investigador, pero no son necesarias para la realización del diagnóstico. Por otro lado, la normalidad de estas otras pruebas, incluyendo el electromiograma, no excluye la existencia de la enfermedad.
Incluso cuando no se conoce en la familia la existencia de DM1, ante un recién nacido hipotónico o, incluso, la detección por ecografía de polihidramnios en el segundo o tercer trimestre de la gestación o escasez de movimientos fetales, se debe considerar la posibilidad de que nos encontremos ante una DM1 y plantear la realización del test genético en el niño y sus progenitores.
Debido a la complejidad de la enfermedad, se recomienda que los pacientes con DM1 sean seguidos en unidades de enfermedades neuromusculares con experiencia en el manejo de la DM1 y con acceso a múltiples especialidades. Cada especialista que participe en la atención debe tener conocimientos adecuados sobre la DM1.
Afectación neurológicaProblemas muscularesDebilidadLa debilidad en la DM1 es de neto predominio distal; también se afectan los músculos del cuello, de la cara, de la masticación, deglución y fonación. Tiene un curso progresivo que se estima en un 1-3% anual5. El riesgo de caídas y de fracturas está aumentado.
Para cuantificar la debilidad se utilizan varias escalas.
- i)
La Muscular Impairment Rating Scale (MIRS)6 es una escala validada para conocer el grado de debilidad muscular. Se evalúan 11 músculos de forma manual con una escala de 1 (sin debilidad) a 5 (importante debilidad). Los grupos musculares son:
- a.
Flexores del cuello
- b.
Abductores del hombro bilateral
- c.
Flexores del codo bilateral
- d.
Extensores de la muñeca bilateral
- e.
Flexores de los dedos bilateral
- f.
Flexores de la cadera bilateral
- g.
Extensión de la rodilla bilateral
- h.
Flexores de la rodilla bilateral
- i.
Dorsiflexores del tobillo bilateral
- j.
Flexión plantar del tobillo bilateral
- ii)
La escala del Medical Research Council (MRC)
- iii)
Utilización de un dinamómetro en la garra manual
- iv)
Test funcionales7:
- •
Marcha en 6min (distancia)
- •
Marcha/carrera de 10m (prueba cronometrada)
- •
Subir 4 escalones (prueba cronometrada)
- •
Bajar 4 escalones (prueba cronometrada)
- •
Levantarse del suelo (prueba cronometrada)
- v)
Debe preguntarse siempre por:
- •
Limitaciones para caminar
- •
Caídas
- •
Limitaciones con las transferencias
- •
Uso y conveniencia de silla de ruedas
El ejercicio aeróbico y el entrenamiento de fuerza de intensidad moderada no provocan daños, aunque no han demostrado utilidad8. El ejercicio moderado se recomienda a todos los pacientes5. Un ejercicio excesivo podría acelerar la progresión de la enfermedad y está desaconsejado. Se ha comprobado que los afectados por DM1 sufren caídas y traspiés hasta 10 veces más que la población general9 con el consiguiente riesgo de fracturas, por lo que se debe valorar la posibilidad de utilizar ortesis antiequino para prevenir estos accidentes10.
MiotoníaLa miotonía es un trastorno de la relajación muscular tras una contracción voluntaria; los pacientes suelen contarlo como rigidez; la miotonía mejora con el calor y con el ejercicio repetido y aumenta con el reposo y el frío. Es un síntoma muy frecuente pero que produce pocos síntomas y que no hay que tratar siempre. Puede favorecer la aparición de rigidez muscular, disartria, disfagia, dolor y síntomas gastrointestinales.
TratamientoLa mexiletina a dosis de 100mg o 200mg 3 veces al día ha demostrado efectividad en la miotonía de la DM1, con evidencia de clase 111. Hay que tener precaución con la utilización de la mexiletina y otros antiarrítmicos en la miotonía por el efecto bloqueante sobre los canales de sodio cardíacos (con alteraciones funcionales en la DM1) ya que actúan sobre la excitabilidad de estos canales, con el consiguiente impacto tanto sobre arritmias cardíacas como sobre la conducción aurículo-ventricular12–15.
Otros tratamientos que se han utilizado son fenitoína, carbamazepina, clomipramina, imipramina, amitriptilina, nifedipino, flecainida, acetazolamida y taurina. Las precauciones con la mexiletina son trasladables a otros fármacos16.
MialgiasEl dolor es un síntoma muy frecuente que puede alcanzar al 90% de los pacientes, por el que se debe preguntar siempre y tratar con analgésicos de manera adecuada17.
TratamientoPuede probarse pregabalina a dosis bajas 50-75mg/día.
FatigaEs un síntoma frecuente en DM y sin clara relación con la debilidad muscular18. No hay escalas específicas para la DM1 validadas en castellano, pero puede utilizarse la escala de intensidad de la fatiga19.
TratamientoSe recomienda modafinilo 200mg/día durante la primera semana, y se puede doblar la dosis en caso de efecto insuficiente20. No obstante, esta indicación está fuera de ficha técnica.
Problemas del sistema nervioso centralLa participación del sistema nervioso central en la DM1 puede llegar a ser uno de los mayores problemas para la vida diaria de los pacientes. Las manifestaciones, sumamente variables, incluyen déficit cognitivo, apatía, fatiga, alteraciones del sueño y, en la DM1 de comienzo neonatal, retraso mental, déficit de atención con hiperactividad y dificultad para funciones ejecutivas21,22.
Retraso intelectualMuy evidente en las formas congénitas, puede observarse también en las otras formas, en grado variable, aunque algunos casos muestran un cociente intelectual normal23. Los pacientes con DM1 tienen un nivel de estudios estadísticamente más bajo que los controles, con una situación socioeconómica inferior, más notable en los varones24.
Se ha demostrado un déficit progresivo de la memoria, incluso en pacientes con una expansión relativamente pequeña25,26.
No existe actualmente un consenso generalizado sobre las pruebas que pueden realizarse para estudiar el déficit cognitivo en la DM1. Se ha propuesto una batería de cribado24, que incluye los siguientes test: 1) Wechsler Adult Intelligence Scale (test de WAIS) breve27 para el cociente intelectual o el correspondiente test en la edad infantil: Weschler Intelligence Scale for Children (WISC) y Weschler Prescholar and Primary Scale Intelligence (WPPSI); 2) Rey's Auditory Verbal Learning Test (test de RAVLT), para evaluar la memoria verbal; 3) la prueba de la figura completa de Rey, que explora la memoria visual; 4) la parte de dígitos y cubos del test de WAISIV, para evaluar la atención y la velocidad de procesamiento. Debe tenerse en consideración, al aplicar estas pruebas, algunos posibles factores de confusión, como es la debilidad, la miotonía, la fatiga, la depresión, etc. Por ello, estos test son de aplicación en casos escogidos o en estudios académicos.
En la forma congénita son frecuentes los trastornos del lenguaje, retraso intelectual, disfunción neurocognitiva, déficit de atención, hiperactividad y trastornos del espectro autista28,29.
Recientemente se ha propuesto que la terapia cognitiva conductual podría mejorar la fatiga y la participación en actividades sociales de los pacientes con DM130.
Trastornos del sueñoSon, a veces, el primer motivo de consulta de la enfermedad. La hipersomnia diurna se manifiesta en un tercio de los casos, en especial, tras las comidas21,31, con dificultades para mantenerse despierto, y se han descrito movimientos periódicos durante el sueño32. Es muy frecuente el síndrome de apnea/hipopnea del sueño (SAHS). Sin embargo, la hipersomnia diurna no se relaciona bien con el SAHS ni mejora siempre con el tratamiento.
El test de Epworth es una buena herramienta para el diagnóstico de estos trastornos33.
Depresión y trastornos de la personalidadAunque los problemas psiquiátricos no suelen ser graves, la depresión mayor y las alteraciones de la personalidad confieren a los pacientes con DM1 unas características psicológicas especiales34. Es frecuente que muestren rasgos de trastorno esquizoide, ansiedad, histeria, compulsión, neurosis depresiva, carácter autodestructivo o narcisista, falta de iniciativa y apatía24,35,36.
La depresión se evalúa habitualmente mediante la escala de Hamilton37.
Enfermedad cerebrovascularLa incidencia de ictus en la DM1 no ha sido bien estudiada y se relaciona con la presencia de arritmias cardioembólicas38–40.
NeuroimagenLos trastornos del sistema nervioso central parecen guardar una cierta correlación con estudios histopatológicos41 y con hallazgos de neuroimagen. Así, se ha demostrado disminución del volumen de la sustancia gris y disrupción difusa de la sustancia blanca, en paralelo con el tamaño de la expansión de tripletes CTG21,42–44.
La FDG-PET puede demostrar hipometabolismo frontotemporal bilateral, aunque no se ha demostrado correlación con el déficit neuropsicológico42.
Afectación cardíacaLa afectación cardíaca es un hecho frecuente e importante en la DM1. Se estima que el 75-80% de los pacientes presenta algún grado de afectación cardíaca con un espectro clínico variable, desde alteraciones leves del electrocardiograma (ECG) hasta arritmias graves que ocasionan muerte súbita. De hecho, hasta un tercio de los fallecimientos en estos pacientes se explican por causas de origen cardíaco.
En un registro nacional danés, que incluyó a 1.186 pacientes con DM1, se comprobó una especial incidencia de arritmias, miocardiopatía e insuficiencia cardíaca45. La base fisiopatológica de estos trastornos parece residir en la sustitución fibroadiposa del tejido especializado de conducción cardíaca y muscular de ambos ventrículos46–48.
Aunque existen datos contradictorios, no se ha podido demostrar una clara relación entre número de repeticiones CTG o la afectación neuromuscular con los eventos cardíacos47. Por ello, y dado que el riesgo de presentar afectación cardíaca es elevado desde edades jóvenes49, es fundamental realizar una detección precoz de dichas complicaciones mediante un seguimiento de por vida, incluso en pacientes sin afectación o con mínima afectación muscular.
Trastornos de conducción, arritmias y muerte súbitaLas manifestaciones cardíacas más frecuentes de la DM1 son las alteraciones electrocardiográficas y las arritmias. El sistema de conducción se afecta prioritariamente a nivel del sistema His-Purkinje, pero también puede hacerlo a otros niveles (sinoauricular y nodo auriculoventricular)5,6. En un metaanálisis reciente47 se señala que los trastornos más comunes son, en orden de frecuencia, los bloqueos auriculoventriculares (BAV) de primer grado (28,2%), la prolongación del intervalo QT (22%) y del complejo QRS (19,9%), la extrasistolia ventricular (14,6%), la fibrilación/flutter auricular (5%), el bloqueo de rama izquierda (5,7%), el bloqueo de rama derecha (4,4%) y la taquicardia ventricular no sostenida (4,1%).
Las arritmias son causantes de palpitaciones, síncope y muerte súbita, aunque en ocasiones el paciente puede permanecer clínicamente asintomático. El riesgo de muerte súbita en pacientes con DM1 se estima en un 0,56% al año47. Existen datos contradictorios sobre cuáles son los mecanismos que subyacen a esta situación. Tradicionalmente se han señalado las bradiarritmias que desencadenan asistolia o fibrilación ventricular como la principal causa de muerte. Es conocido que los pacientes asintomáticos con DM1 y con un intervalo His-ventricular (HV)≥70ms, medidos en un estudio electrofisiológico (EEF) tienen un riesgo elevado de desarrollar un BAV completo y se beneficiarían del implante de un marcapasos profiláctico50. Sin embargo, parecen existir otras causas de muerte súbita no evitables con marcapasos50. Las taquiarritmias, en especial las ventriculares, se han referido como causa de muerte súbita51.
En el estudio de Groh et al. se identificaron como predictores de muerte súbita los antecedentes de taquiarritmias auriculares y la existencia de alteraciones graves del ECG definidas como BAV de segundo o tercer° grado, un PR≥240ms o un QRS≥120ms52. Más recientemente se ha publicado un estudio retrospectivo de una cohorte de pacientes con DM1 y alteración basal del ECG (PR>200ms y/o QRS>100ms), en el que una estrategia invasiva con EEF e implantación de marcapasos profiláctico a pacientes con HV≥70ms se asociaba con una mayor supervivencia durante el seguimiento con respecto a quienes se les realizaba una estrategia no invasiva53.
Afectación miocárdicaEn un porcentaje variable de pacientes con DM1 (7-20%) se ha observado hipertrofia, dilatación y disfunción sistólica de ventrículo izquierdo47,53. Una mayor edad, el sexo masculino, las anomalías de conducción del ECG y la existencia de arritmias son predictores de disfunción sistólica54. Esta entidad, que inicialmente suele ser subclínica, aumenta el riesgo de muerte súbita. Asimismo, se ha descrito una mayor prevalencia de no compactación del ventrículo izquierdo (VI) y de fibrosis miocárdica detectable por realce miocárdico tardío en la RMN55. Por otra parte, en pacientes con enfermedad de Steinert se ha referido una reducción de la masa miocárdica y disfunción del ventrículo derecho, así como una elevada prevalencia del prolapso de la válvula mitral. Finalmente, debe considerarse la existencia de alteraciones en la relajación miocárdica análogas a la miotonía del músculo esquelético que pueden manifestarse en síntomas de insuficiencia cardíaca48.
Recomendaciones56,57 (tabla 3):
- 1.
Los pacientes con DM1, incluso los asintomáticos y sin datos de afectación cardíaca, deben hacer seguimiento cardiológico de por vida, en busca de posibles complicaciones, dado el carácter progresivo de la enfermedad.
- 2.
Los pacientes con DM1 deben recibir información acerca de los síntomas de alarma (síncope, palpitaciones) por los que deben consultar de forma urgente.
- 3.
Se recomienda realizar anualmente una anamnesis dirigida (con especial atención a la existencia de síncope y/o palpitaciones), así como ECG y holter de ECG.
- 4.
Se recomienda realizar un ecocardiograma transtorácico cada 3-5 años.
- 5.
El manejo de las alteraciones estructurales cardíacas es similar al de la población general.
- 6.
Los pacientes asintomáticos con alteraciones significativas del ECG (PR>200ms, QRS>100ms) o los que tienen antecedentes de síncope de perfil cardiogénico podrían beneficiarse de la realización de un EEF invasivo para medir los tiempos de conducción y el riesgo de inducción de arritmias ventriculares. En el caso de pacientes con un EEF normal, debe considerarse repetirlo durante el seguimiento en caso de aparición de nuevos síntomas o progresión significativa de los trastornos del ECG basal (al menos un incremento>20ms del PR y/o QRS).
- 7.
Los pacientes con HV>70ms en EEF o BAV de segundo grado en ECG, por su elevada tasa de progresión a BAV completo, se benefician del implante profiláctico de un marcapasos definitivo. El marcapasos no previene completamente la muerte súbita, puesto que esta puede producirse por otros mecanismos (arritmias ventriculares, asistolia).
- 8.
Se debe considerar el implante de desfibrilador automático implantable en los pacientes con DM1 y antecedentes de arritmias ventriculares y/o disfunción del VI. En pacientes con taquicardia ventricular monomórfica sostenida debe realizarse un EEF y, en caso de comprobarse un mecanismo de reentrada rama-rama, debe realizarse ablación con catéter.
- 9.
El manejo de las arritmias supraventriculares en pacientes con DM1 es similar al de otros grupos de pacientes. Sin embargo, debe tenerse especial precaución en el uso de fármacos antiarrítmicos que puedan prolongar el HV (especialmente los del grupo i).
- 10.
La sedación para cardioversión eléctrica en pacientes con DM1 debe realizarse con precaución en un medio adecuado y por personal entrenado, por el riesgo de fracaso respiratorio.
Recomendaciones cardiológicas
| 1. Los pacientes con DM1, incluso los asintomáticos y sin datos de afectación cardíaca, deben recibir seguimiento cardiológico de por vida, en busca de posibles complicaciones, dado el carácter progresivo de la enfermedad |
| 2. Los pacientes con DM1 deben recibir información acerca de los síntomas de alarma (síncope, palpitaciones) por los que deben consultar de forma urgente |
| 3. Se recomienda realizar anualmente una anamnesis dirigida (especial atención a la existencia de síncope y/o palpitaciones), así como ECG y holter de ECG |
| 4. Se recomienda realizar un ecocardiograma transtorácico cada 3-5 años |
| 5. Las alteraciones estructurales cardíacas se manejarán como en la población general |
| 6. Los pacientes asintomáticos con alteraciones significativas del ECG (PR>200ms, QRS>100ms) o los que tienen antecedentes de síncope de perfil cardiogénico podrían beneficiarse de la realización de un EEF invasivo para medir los tiempos de conducción y el riesgo de inducción de arritmias ventriculares. En el caso de pacientes con un EEF normal, debe considerarse repetirlo durante el seguimiento en caso de aparición de nuevos síntomas o progresión significativa de los trastornos del ECG basal (al menos un incremento>20ms del PR y/o QRS) |
| 7. Los pacientes con HV>70ms en EEF o BAV de segundo grado en ECG, por su elevada tasa de progresión a BAV completo, se benefician del implante profiláctico de un marcapasos definitivo. El marcapasos no previene completamente la muerte súbita, puesto que existen otros mecanismos (arritmias ventriculares, asistolia) |
| 8. Se debe considerar el implante de desfibrilador automático implantable en los pacientes con DM1 y antecedentes de arritmias ventriculares y/o disfunción del VI. En pacientes con taquicardia ventricular monomórfica sostenida debe realizarse un EEF y, en caso de comprobarse un mecanismo de reentrada rama-rama, debe realizarse ablación con catéter |
| 9. El manejo de las arritmias supraventriculares en pacientes con DM1 es similar al de otros grupos de pacientes; sin embargo, debe tenerse especial precaución en el uso de fármacos antiarrítmicos que puedan prolongar el HV (especialmente los del grupo i) |
| 10. La sedación para cardioversión eléctrica en pacientes con DM1 debe realizarse con precaución en un medio adecuado y por personal entrenado, por el riesgo de fracaso respiratorio de estos pacientes |
La afectación respiratoria en la DM1 es frecuente y constituye una de las principales causas de muerte prematura en estos pacientes: llega a explicar el 51-75% de los fallecimientos58,59. Es, además, uno de los factores que más influyen en el deterioro de la calidad de vida1. Parece existir una cierta correlación entre el tamaño de la expansión y la intensidad de la afectación respiratoria60,61. Con respecto a los factores de riesgo de insuficiencia respiratoria, algunos estudios muestran un mayor riesgo en los hombres62. Además, la obesidad, frecuente en la DM1, ha demostrado ser un factor de riesgo independiente de fallo respiratorio63.
En la mayoría de los casos la afectación respiratoria es de instauración insidiosa y progresiva, por lo que muchas veces se diagnostica con retraso, al pasar desapercibida; ocurre tardíamente (entre los 50-60 años) y en pacientes con diagnóstico previo ya establecido de DM1, con sintomatología muscular y multisistémica64. Sin embargo, hay casos en los que la insuficiencia respiratoria puede aparecer de forma aguda, desencadenada por diferentes procesos como puede ser un procedimiento anestésico o una infección respiratoria65.
Los mecanismos fisiopatogénicos de la alteración respiratoria en estos enfermos continúan sin ser bien conocidos, pero son muchos los estudios que apoyan la hipótesis de un doble mecanismo: periférico (por la afectación muscular distrófica y miotónica de los músculos implicados en la respiración [diafragma y musculatura abdominal e intercostal]) y central (por anomalías del sistema nervioso central que condicionan una alteración del control central de la respiración)66,67.
La afectación respiratoria de los pacientes con DM1 puede estar relacionada con alteración ventilatoria restrictiva debida a la debilidad muscular o con la presencia de SAHS o hipoventilación nocturna. Por tanto, las manifestaciones clínicas más frecuentes que sugieren una afectación respiratoria son las infecciones respiratorias recurrentes, la disnea progresiva, la hipersomnolencia diurna y la cefalea matutina66,68–71.
La afectación conductual y cognitiva de los pacientes con DM1 puede dificultar la correcta identificación de los síntomas respiratorios y una mala adhesión a las terapias nocturnas respiratorias72.
La debilidad de los músculos respiratorios que provoca una alteración ventilatoria restrictiva ocasiona hipoventilación, inicialmente solo nocturna.
La debilidad de los músculos espiratorios (intercostales y abdominales) aumenta el riesgo de infecciones respiratorias por disminución de la capacidad para toser y por la disfagia. El riesgo de neumonía se ve además incrementado por un mayor riesgo de broncoaspiración debido a la debilidad orofaríngea y esofágica73,74.
La heterogeneidad fisiopatogénica y clínica de la afectación respiratoria en los pacientes afectos de DM1 obliga a protocolizar los estudios de cribado de esta complicación.
Recomendaciones (tabla 4):
- 1.
La evaluación de la disfunción respiratoria en los pacientes con DM1 debe ser periódica.
- 2.
La anamnesis respiratoria debe incluir síntomas de disfagia, eficacia de la tos, disnea, hipersomnia diurna, cefalea y cansancio matutino. Se recomienda la escala de somnolencia de Epworth33.
- 3.
El estudio inicial debe incluir espirometría, pletismografía, estudio de presiones espiratoria e inspiratoria máximas (PIM y PEM) o SNIF, pico de tos (PECF), gasometría basal y poligrafía cardiorrespiratoria nocturna (PGR).
- 4.
La polisomnografía nocturna (PSG) se recomienda en caso de hipersomnia diurna con PGR normal o patológica y sin respuesta a la terapia ventilatoria nocturna.
- 5.
Se recomienda tratamiento con CPAP si SAHS moderado o grave y ausencia de hipoventilación nocturna (CT90>30%).
- 6.
Se recomienda tratamiento con ventilación mecánica no invasiva (BiPAP) si predomina la hipoventilación nocturna (CT90>30%).
- 7.
Se recomienda un entrenamiento inicial intra- o extrahospitalario de los dispositivos nocturnos para mejorar la adherencia al tratamiento.
- 8.
Se recomienda tratamiento antibiótico precoz en el caso de infecciones respiratorias.
- 9.
Se recomienda, para el manejo de secreciones, realizar técnicas de fisioterapia respiratoria y el uso de asistentes mecánicos para la tos con pautas de in/exsuflación (PECF<270).
- 10.
Vacunación anual de la gripe y vacunación frente al neumococo al menos una vez en la vida.
- 11.
Antes de cualquier cirugía con anestesia general debe realizarse una evaluación respiratoria.
Recomendaciones neumológicas
| 1. La evaluación de la disfunción respiratoria en los pacientes con DM1 debe ser periódica |
| 2. La anamnesis respiratoria debe incluir síntomas de disfagia, eficacia de la tos, disnea, hipersomnia diurna, cefalea y cansancio matutino. Se recomienda la escala de somnolencia de Epworth33 |
| 3. El estudio inicial debe incluir espirometría, pletismografía, estudio de presiones espiratoria e inspiratoria máximas (PIM y PEM) o SNIF, pico de tos (PECF), gasometría basal y poligrafía cardiorrespiratoria nocturna (PGR) |
| 4. La polisomnografía nocturna (PSG) se recomienda en caso de hipersomnia diurna con PGR normal o patológica y sin respuesta a la terapia ventilatoria nocturna |
| 5. Se recomienda tratamiento con CPAP si SAHS moderado o grave y ausencia de hipoventilación nocturna (CT90>30%) |
| 6. Se recomienda tratamiento con ventilación mecánica no invasiva (BiPAP) si predomina la hipoventilación nocturna (CT90>30%) |
| 7. Se recomienda un entrenamiento inicial intra- o extrahospitalario de los dispositivos nocturnos para mejorar la adherencia al tratamiento |
| 8. Se recomienda tratamiento antibiótico precoz en el caso de infecciones respiratorias |
| 9. Se recomienda para el manejo de secreciones realizar técnicas de fisioterapia respiratoria y el uso de asistentes mecánicos para la tos con pautas de in/exsuflación (PECF<270). |
| 10. Vacunación anual de la gripe y vacunación frente al neumococo al menos una vez en la vida |
| 11. Antes de cualquier cirugía con anestesia general debe realizarse una evaluación respiratoria |
La DM1 puede afectar a múltiples órganos y tejidos, en grado variable.
DermatologíaPilomatricomas múltiples (epitelioma calcificante de Malherbe)Es un tumor benigno derivado de las células primitivas de la matriz del folículo piloso que se manifiesta en forma de nódulos subcutáneos, no dolorosos, de 0,5-5cm de diámetro. Se localiza en cuero cabelludo, cara, cuello y miembros superiores. Puede tratarse mediante exéresis quirúrgica, cuando existe una molestia local o problema estético y el diagnóstico histopatológico es confirmatorio. Puede confundirse con quistes sebáceos simples75,76.
AlopeciaEs de causa multifactorial, por envejecimiento acelerado cutáneo y del folículo piloso y por cambios hormonales. No existen recomendaciones terapéuticas específicas77.
Dermatitis seborreicaSe correlaciona con valores disminuidos de vitamina D sérica. El diagnóstico se lleva a cabo por exploración dermatológica y análisis de vitamina D sérica. Puede mejorar con calcifediol6.
Nevus displásico, no asociado a melanomaSe asocia a valores disminuidos de vitamina D sérica. El diagnóstico se realiza por exploración dermatológica y se completa por análisis séricos de vitamina D. Puede mejorar con calcifediol77.
Endocrinología y metabolismoLos pacientes con DM1 pueden presentar diferentes endocrinopatías, siendo las más prevalentes el hipogonadismo, las alteraciones tiroideas, las anomalías del metabolismo hidrocarbonado y las del fosfocálcico. Con menor frecuencia pueden presentar disfunción del eje corticotropo, dislipidemia y alteraciones electrolíticas. La incidencia de patología endocrinológica aumenta con la evolución de la enfermedad, y por ello es importante el cribado periódico78.
Hipogonadismo hipergonadotropoEs la manifestación endocrinológica más frecuente en la DM. Se ha descrito que hasta el 80% de los varones presentan atrofia testicular, con mayor daño tubular que intersticial. Se caracteriza por niveles disminuidos de testosterona y/o infertilidad, por afectación de la espermatogénesis y elevación de gonadotropinas (hormona folículo-estimulante [FSH], hormona luteinizante [LH]). El déficit de testosterona produce una regresión progresiva de los caracteres sexuales secundarios (disminución del vello corporal, de la libido y de la frecuencia de afeitado) y menor masa muscular. Esta disminución de la masa magra puede a su vez empeorar la sintomatología muscular de base de la enfermedad, y a largo plazo puede inducir menor masa ósea.
En las mujeres puede existir más incidencia de infertilidad, abortos espontáneos y, en casos raros, fallo ovárico precoz. El fallo ovárico precoz es, además, causa de osteoporosis secundaria79.
Dada la alta prevalencia de hipogonadismo, se recomienda solicitar LH/FH y testosterona a los varones en edad adulta en la primera visita y posteriormente con frecuencia anual o antes, si presentaran clínica de hipogonadismo.
En las mujeres con amenorrea secundaria, se debe solicitar estradiol y LH/FSH.
Si existe hipogonadismo masculino, debe remitirse a endocrinología para completar estudio y valorar, ofrecer tratamiento sustitutivo con testosterona.
En las mujeres, dependiendo de la edad de presentación del fallo ovárico, se valorará terapia hormonal sustitutiva.
Alteraciones del metabolismo hidrocarbonadoEn la DM1, el defecto fisiopatológico subyacente de las alteraciones del metabolismo hidrocarbonado es generalmente la resistencia a la insulina, por una menor sensibilidad de la misma a nivel muscular. Puede manifestarse como:
- -
Hiperinsulinismo con tolerancia normal a la glucosa: niveles elevados de insulina con glucemia plasmática normal.
- -
Prediabetes que incluye glucemia basal alterada (glucemia plasmática en ayunas 100-125mg/dl), hemoglobina glucosilada (HbA1c): 5,7-6,4% (método estandarizado por el Diabetes Control and Complications Trial [DCCT]-National Glycohemoglobin Standardization Program [NGSP]) o intolerancia a hidratos de carbono (glucemia a las 2h tras sobrecarga oral de 75g de glucosa de 140-199mg/dl).
- -
Diabetes mellitus diagnosticada por glucemia plasmática en ayunas≥126mg/dl, glucemia tras sobrecarga oral con 75g de glucosa≥200mg/dl o HbA1c≥6,5%.
Para el diagnóstico de prediabetes o diabetes, se requieren al menos 2 criterios salvo que el paciente presente una glucemia plasmática al azar mayor de 200mg/dl con manifestaciones clínicas (poliuria, polidipsia, polifagia, pérdida ponderal).
Se aconseja determinar glucemia plasmática en ayunas, HbA1c, e insulina una vez al año o antes si hubiera sintomatología cardinal de hiperglucemia: poliuria, polidipsia, polifagia, pérdida de peso.
El tratamiento antidiabético de elección en los pacientes con diabetes mellitus son los fármacos sensibilizadores de la insulina. La metformina es el de primera elección.
Alteraciones lipídicasLas alteraciones lipídicas más frecuentes son la elevación de los triglicéridos y el descenso del HDL presentes en el 67% y el 35% de los pacientes respectivamente. La dislipidemia, junto con las alteraciones del metabolismo hidrocarbonado, predispone a un mayor riesgo cardiovascular debido al síndrome metabólico, por lo que se debe vigilar la presión arterial y el perfil lipídico (colesterol y sus fracciones y triglicéridos) 1-2 veces al año.
Es fundamental insistir en realizar una alimentación saludable y ejercicio físico habitual, adaptado a cada individuo, puesto que el sedentarismo de estos pacientes es el factor etiopatogénico fundamental en la aparición del síndrome metabólico. En caso de necesitar tratamiento farmacológico para la dislipidemia, podrían utilizarse estatinas y fibratos, pero con vigilancia estrecha, debido a que podrían empeorar la clínica de miopatía de estos pacientes. Si se prescriben, debe monitorizarse la CK, la transaminasa glutámico-oxalacética (GOT) y la transaminasa glutámico pirúvica (GPT) periódicamente y sobre todo durante el primer año80.
Metabolismo fosfocálcicoLa prevalencia de déficit de vitamina D, definido como un valor menor de 30ng/ml, llega en algunas series a detectarse en el 90% de los pacientes con DM1. El déficit grave (nivel de 25 OH vitamina D<10ng/ml) se detecta en el 40%. Las causas de la hipovitaminosis D son las mismas que se describen para la población general: baja ingesta de alimentos ricos en esta vitamina, poca exposición solar y los cambios en la composición corporal (incremento de la masa grasa). El déficit de vitamina D puede ocasionar osteomalacia (insuficiente mineralización del hueso), debilidad muscular e hiperparatiroidismo secundario, que a su vez empeora la debilidad muscular ya presente en los sujetos con distrofia miotónica.
En relación con el metabolismo fosfocálcico, existe una mayor prevalencia de hiperparatiroidismo primario, hasta en un 17,5%, generalmente asociado a adenomas paratiroideos78.
El manejo del hiperparatiroidismo primario en los pacientes con DM1 es el mismo que en la población general.
Se recomienda vigilar los niveles de calcio, fósforo, vitamina D y parathormona (PTH) una vez al año. En caso de hipercalcemia, hipofosforemia o niveles de PTH elevados, debe solicitarse una segunda determinación y si se confirma la alteración remitir a endocrinología.
Metabolismo tiroideoLas alteraciones tiroideas son menos frecuentes que el resto de endocrinopatías, a diferencia de lo que acontece en el resto de la población. La patología nodular es la objetivada con mayor frecuencia en los pacientes con DM1, y en la mayoría cursa con eutiroidismo.
En caso de hipotiroidismo primario, la clínica característica de hipofunción hormonal puede incrementar la debilidad muscular y empeorar los síntomas de la distrofia, por lo que es importante su diagnóstico precoz y tratamiento sustitutivo.
Se recomienda monitorizar la función tiroidea (tirotropina [TSH]) y exploración física de la región tiroidea una vez al año. Si se objetiva crecimiento del tiroides o se detecta nódulo tiroideo a la exploración física, debe realizarse ecografía tiroidea y derivar al endocrinólogo, así como si se evidencia alteración de TSH en 2 determinaciones81–83.
Eje corticotropoOtra de las anomalías endocrinológicas observadas en los pacientes con DM1 son las alteraciones del eje corticotropo (hormona liberadora de corticotropina [CRH]-corticotropina [ACTH]-cortisol) con disregulación adrenocortical, cuya frecuencia no está claramente establecida. Los resultados obtenidos hasta ahora son variables, así como su significación clínica. Podría existir una hiperactividad, reflejada por aumento de cortisol basal y ACTH, con un ritmo circadiano aplanado de cortisol y ACTH. La respuesta de cortisol a la estimulación de ACTH exógena es variable. En la mayoría de los pacientes responde el cortisol plasmático adecuadamente, aunque hay algún caso de hiporrespuesta, lo que implicaría una baja reserva adrenal, y en pocos casos hiperrespuesta, en presencia de mayores repeticiones de CTG. Tras CRH se aprecia una respuesta exagerada de ACTH.
Otros autores, por el contrario, han hallado cifras bajas de cortisol basal, y tras estímulo de CRH menor respuesta de cortisol, con cifras medias de ACTH más altas, lo que indica una falta de eficacia de ACTH sobre el receptor adrenal.
Las pruebas de estimulación con insulina y metirapona son normales, así como las de supresión con dexametasona84–86.
No es necesario evaluar este eje de forma rutinaria, salvo que existan manifestaciones clínicas de hipocortisolismo (astenia, hipotensión ortostática, hiperpotasemia o hiponatremia), para lo cual habría que solicitar cortisol basal en sangre a las 8.00, y si es menor de 18μg/dl, remitir a endocrinología. También sería recomendable la derivación a endocrinología a los pacientes con clínica de hipercortisolismo (obesidad central, fragilidad capilar, plétora facial, estrías rojizas en abdomen…). Para el cribado de secreción aumentada de cortisol se solicita cortisol libre en orina de 24h, cortisol salival o supresión con un miligramo de dexametasona a las 23.00 horas con determinación de cortisol a las 8.00 horas. Si el cortisol en sangre tras supresión es mayor de 1,8μg/dl, la cortisoluria o el cortisol salival están elevados, derivar a endocrinología.
Se han descrito asimismo casos aislados de alteraciones hidroelectrolíticas, cuyas causas aún no están claramente establecidas. Algunos pacientes presentan hiperpotasemia, que parece secundario a un hipoaldosteronismo hiperreninémico87 y en otros casos se han detectado cifras elevadas de sodio, sin repercusión clínica, que se postula que puedan deberse a una alteración de la osmorregulación del sodio secundaria a déficit de la producción de ADH y disminución de la sed88.
Se recomienda solicitar anualmente iones plasmáticos, y si están alterados, repetirlos ampliando con determinación de sodio y potasio en orina. En caso de persistir con alteraciones electrolíticas, deberán ser remitidos a endocrinología para su estudio.
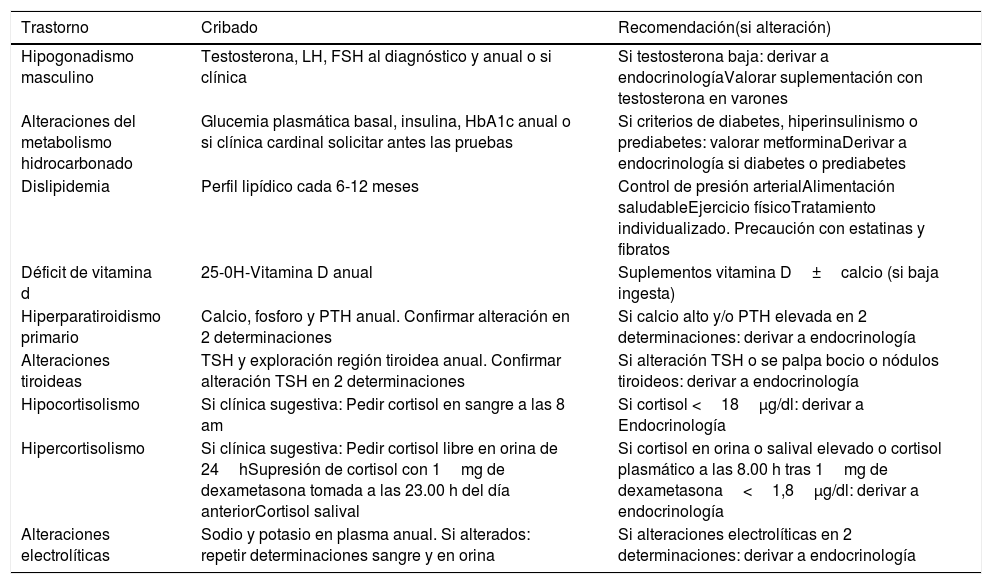
En la tabla 5 se recogen las recomendaciones sobre el seguimiento endocrinológico.
Recomendaciones endocrinológicas
| Trastorno | Cribado | Recomendación(si alteración) |
|---|---|---|
| Hipogonadismo masculino | Testosterona, LH, FSH al diagnóstico y anual o si clínica | Si testosterona baja: derivar a endocrinologíaValorar suplementación con testosterona en varones |
| Alteraciones del metabolismo hidrocarbonado | Glucemia plasmática basal, insulina, HbA1c anual o si clínica cardinal solicitar antes las pruebas | Si criterios de diabetes, hiperinsulinismo o prediabetes: valorar metforminaDerivar a endocrinología si diabetes o prediabetes |
| Dislipidemia | Perfil lipídico cada 6-12 meses | Control de presión arterialAlimentación saludableEjercicio físicoTratamiento individualizado. Precaución con estatinas y fibratos |
| Déficit de vitamina d | 25-0H-Vitamina D anual | Suplementos vitamina D±calcio (si baja ingesta) |
| Hiperparatiroidismo primario | Calcio, fosforo y PTH anual. Confirmar alteración en 2 determinaciones | Si calcio alto y/o PTH elevada en 2 determinaciones: derivar a endocrinología |
| Alteraciones tiroideas | TSH y exploración región tiroidea anual. Confirmar alteración TSH en 2 determinaciones | Si alteración TSH o se palpa bocio o nódulos tiroideos: derivar a endocrinología |
| Hipocortisolismo | Si clínica sugestiva: Pedir cortisol en sangre a las 8 am | Si cortisol <18μg/dl: derivar a Endocrinología |
| Hipercortisolismo | Si clínica sugestiva: Pedir cortisol libre en orina de 24hSupresión de cortisol con 1mg de dexametasona tomada a las 23.00 h del día anteriorCortisol salival | Si cortisol en orina o salival elevado o cortisol plasmático a las 8.00 h tras 1mg de dexametasona<1,8μg/dl: derivar a endocrinología |
| Alteraciones electrolíticas | Sodio y potasio en plasma anual. Si alterados: repetir determinaciones sangre y en orina | Si alteraciones electrolíticas en 2 determinaciones: derivar a endocrinología |
La afectación del aparato digestivo es una de las más frecuentes y no ha sido bien estudiada. Es habitual que las manifestaciones derivadas de su alteración sean poco valoradas por paciente y clínico. Se debe fundamentalmente a la afectación del músculo liso en la DM1. La frecuencia e intensidad de los síntomas que provoca son muy variables, si bien pueden llegar a ser graves e, incluso, ser la forma de presentación de la enfermedad. Se ha atribuido este trastorno a inclusiones de ARN y de proteína «muscleblind-like 1» (MBNL1) en los núcleos del músculo liso89. Otras hipótesis sugieren una deficiente inervación del músculo liso, infiltración grasa de las paredes de las vísceras huecas, fibrosis de estas paredes o, incluso, degeneración del músculo liso. Para estas hipótesis no hay, sin embargo, estudios concluyentes90–93.
Alteraciones en la masticaciónSon producidas por debilidad y miotonía de la musculatura oral y masticatoria (lengua, paladar, faringe, maseteros, pterigoideos). Deben evaluarse clínicamente una vez al año. Si existe afectación, es recomendable fisioterapia con logopeda5.
DisfagiaLa disfagia está presente en aproximadamente el 55% de los pacientes con distrofia miotónica, acarrea un alto riesgo de neumonía por aspiración y desnutrición, y constituye una importante causa de morbimortalidad92.
No se relaciona con el grado de afectación del músculo esquelético ni con la duración de la enfermedad muscular. Se produce por debilidad de músculos masticatorios, músculos faríngeos, esfínter esofágico superior y cuerpo esofágico, así como miotonía de dichos músculos. Presentan menor propulsión faríngea con incremento de residuo a dicho nivel. Todos estos mecanismos con la debilidad de los músculos respiratorios favorecen la aparición de infecciones respiratorias93. La disfagia del paciente con distrofia miotónica tiene la peculiaridad de que suele ocasionar más dificultad para tragar alimentos sólidos, a diferencia de otras enfermedades neurológicas, en las que la dificultad es para alimentos de textura líquida o mezcla de dobles texturas. En todos los enfermos es recomendable realizar una historia dirigida, insistiendo en la presencia de síntomas sugestivos de disfagia como son: tos con la deglución, infecciones respiratorias, y pérdida de peso79. Un test sencillo de cribado que nos puede ayudar a detectar la presencia de disfagia es el Eating Assessment Tool-10 (EAT-10)94. Se trata de un cuestionario de 10 preguntas, validado en español. Si el test es positivo, es necesario realizar diagnóstico de disfagia, para lo que se requieren técnicas realizadas por personal entrenado: test de volumen viscosidad (MECV-V), fibroendoscopia de la deglución o videofluoroscopia. El primero consiste en dar al paciente diferentes volúmenes y texturas y controlar signos de alarma: tos, desaturación, cambios en la voz. De esta manera sabremos la textura y el volumen que son seguros. La fibroendoscopia es un método directo realizado por el especialista en otorrinolaringología, que permite ver si existe penetración o aspiraciones al dar diferentes volúmenes y texturas de alimentos, y al ser un método directo, permite detectar aspiraciones silentes que pueden pasar inadvertidas con el MECV95. Por último, la videofluoroscopia, considerada el patrón oro, es una técnica radiológica en tiempo real con la que podemos estudiar la deglución y su seguridad. Sin embargo, es una técnica costosa y no está disponible en todos los centros.
Para el tratamiento se adaptará la alimentación a la textura más segura en cada caso. Si la disfagia es a líquidos se emplearán espesantes y se evitarán dobles texturas, y si es a sólidos se probará con alimentos blandos o triturados y se evitarán los alimentos de más riesgo para cada paciente. La fisioterapia guiada por un logopeda puede mejorar la clínica.
No hay que olvidar que la disfagia va asociada a riesgo de desnutrición, por lo que es importante vigilar el peso de estos pacientes, para detectar pérdida de peso involuntaria o disminución de la ingesta, así como monitorizar parámetros analíticos de desnutrición: albúmina y prealbúmina. En caso de desnutrición o pérdida de peso, será necesario enriquecer la dieta desde el punto de vista calórico y proteico; si no es suficiente, añadir suplementos nutricionales, y en algunos casos será preciso nutrición enteral mediante sonda nasogástrica, si se prevé que se necesitará poco tiempo (<4-6 semanas), u ostomías (gastrostomías o yeyunostomías) cuando se espera que el trastorno persista.
Además, en el enfoque nutricional debemos incluir la prescripción de ejercicio físico adaptado a cada paciente para preservar o minimizar la pérdida de masa muscular.
Recomendaciones sobre la disfagia:
- 1.
Debe monitorizarse la presencia de síntomas de disfagia a sólidos, líquidos y doble textura al menos una vez al año.
- 2.
Se recomienda el empleo de test de cribado de disfagia EAT-10 una vez al año.
- 3.
Si el test EAT-10 muestra un resultado≥3, realizar test de volumen-viscosidad, fibroendoscopia de la deglución o videofluoroscopia, según disponibilidad del hospital.
- 4.
Vigilar el peso una vez al año o con mayor frecuencia si el paciente presenta disfagia.
- 5.
En los pacientes con disfagia se proporcionarán recomendaciones nutricionales específicas (anexo 1).
- 6.
En los pacientes con desnutrición o pérdida ponderal no intencionada se recomiendan pautas nutricionales de enriquecimiento de alimentación y el empleo de suplementos nutricionales si la pérdida ponderal es>10% en 6 meses o no hay mejoría con las recomendaciones nutricionales.
- 7.
Se fomentará la realización de ejercicio físico individualizado en todas las visitas.
- 8.
De forma similar a otras enfermedades neurológicas o situaciones de disfagia, en los casos en que esta sea grave y no sea posible instaurar recomendaciones específicas, se colocará sonda nasogástrica o gastrostomía según la duración de la situación.
Se han descrito trastornos atribuidos a miotonía esofágica, hipotonía del esófago, debilidad del cardias y a pobreza del peristaltismo esofágico y gástrico.
Puede demostrarse este retraso del tránsito a través del esófago por cinerradiografía, manometría esofágica o, de forma mucho más sencilla, rápida y barata, mediante el estudio del tránsito esofágico con radioisótopos, técnica que ha demostrado una excelente correlación con la manometría esofágica96–99.
El vaciamiento gástrico está enlentecido, lo que se manifiesta con digestiones pesadas y prolongadas y sensación de plenitud abdominal. Esta alteración, unida a debilidad del cardias, favorece también la regurgitación y el riesgo de broncoaspiración. Puede ser tratada con inhibidores de la bomba de protones y procinéticos de musculatura lisa79. El estudio del vaciamiento gástrico con radioisótopos permite confirmar esta alteración100.
IntestinoLa debilidad y escasez de los movimientos peristálticos de intestino delgado y grueso causan fases de estreñimiento alternadas con diarreas, así como molestias abdominales de intensidad variable, que pueden llegar a la seudoobstrucción intestinal. Se han descrito tanto megacolon como vólvulos intestinales asociados a DM1. En los estudios radiológicos puede encontrarse dilatación segmentaria y desaparición de las haustras colónicas, por disminución del peristaltismo100–103.
Se recomienda evaluar anualmente el estreñimiento. Mejora con dieta rica en fibra, procinéticos y laxantes5.
La diarrea es debida al sobrecrecimiento bacteriano intestinal y suele acompañarse de hinchazón abdominal, meteorismo y malestar. Debe vigilarse el posible déficit nutricional, especialmente el de vitamina B12. El diagnóstico es clínico. Existe un test de metabolismo de hidratos de carbono, el test de D-xilosa, en desuso. Debe tratarse con dieta, probióticos, colestiramina y antibióticos (rifaximina 400mg/12h durante 7 días en ciclos mensuales o ciprofloxacino 500mg/12h durante 7 días en ciclos mensuales)104.
Vesícula biliarLa incidencia de colelitiasis está aumentada en pacientes con DM1 (25-50% de los casos), debida a la lenta movilidad de la vesícula, que favorece el aumento del barro biliar y la formación de cálculos105.
AnoEl esfínter anal interno puede mostrar intensa miotonía y el externo una mezcla de miotonía, más leve, y debilidad, lo que contribuye al estreñimiento del paciente. La miotonía puede ponerse de manifiesto por electromiografía106.
Sistema genitourinarioAdemás de las alteraciones hormonales señaladas con anterioridad, y que cursan con hipogonadismo, disminución de la potencia sexual, reducción de la libido e hipofertilidad, hay otros síntomas que pueden aparecer durante el curso de la enfermedad, por afectación del músculo liso.
Así, durante el parto, las mujeres presentan contracciones uterinas inadecuadas, retraso en la relajación uterina y aumento del trabajo de parto. Tras el parto existe un mayor riesgo de hemorragia y de retención de la placenta por incompetencia muscular uterina34.
La vejiga de los pacientes con DM1 suele ser normal, y ocasionalmente pueden encontrarse dilataciones focales en los uréteres34.
EstomatologíaCaries, gingivitis, placa bacterianaLas alteraciones dentarias son especialmente frecuentes a causa de una higiene oral deficiente, en especial en la arcada dentaria posterior, y por hiposalivación. Debe ser valorada anualmente e insistirse en la adecuada higiene oral, con instrucciones para el lavado de los dientes, uso de seda dental, enjuagues con clorhexidina y evitar alimentos azucarados que favorecen la caries107,108.
Dismorfias faciales, paladar ojival, retrognatia o prognatismo mandibularSe encuentran en la DM1 congénita, por alteraciones en el desarrollo embrionario. Si las alteraciones son graves, y alteran la alimentación o el lenguaje, se puede plantear cirugía ortognática109.
Limitación de la apertura bucal, dolor y claudicación mandibulares durante la masticaciónEstas alteraciones se deben a disfunción temporomandibular por cambios morfológicos del disco articular con remodelación ósea y debilidad de musculatura masticatoria. Se estudian con pruebas de imagen de la articulación temporomandibular (radiografía funcional, RMN). Si cursan con dolor, pueden tratarse con placa de descarga intraoral o fisioterapia110.
OftalmologíaCataratasA lo largo de la evolución de la DM1 es sumamente frecuente el desarrollo de cataratas; se trata de unas cataratas peculiares, subcapsulares, iridiscentes, muy características. Se encuentran en el 90% de los pacientes y suelen diagnosticarse en general a partir de los 50 años de edad, aunque pueden aparecer antes. En los casos con 50 a 100 repeticiones del triplete, la aparición de cataratas precoces suele ser el único dato de la enfermedad. Todos los pacientes con DM1 deben ser evaluados periódicamente por un oftalmólogo, al menos cada 2 años.
La exploración oftalmológica, incluyendo la lámpara de hendidura, es el procedimiento diagnóstico habitual. Las cataratas se tratan mediante la extirpación del cristalino111–114.
Hipotensión ocularLa casi totalidad de los pacientes con DM1 tiene hipotensión ocular y el glaucoma es excepcional. Se atribuye a debilidad de la musculatura lisa o a desprendimiento del cuerpo ciliar. Cursa generalmente de forma asintomática, y se diagnostica midiendo la presión intraocular, que suele estar por debajo de 5mmHg. No requiere tratamiento, a menos que condicione cambios funcionales o estructurales en el ojo115.
Ptosis palpebralLa ptosis palpebral, por debilidad del músculo elevador del párpado, suele ser constante en esta enfermedad. Tiene un curso progresivo y contribuye al aspecto característico de la cara de estos pacientes. Si llega a limitar el campo visual debe plantearse blefaroplastia116.
EmbarazoLas pacientes con DM1, embarazadas, tienen riesgo de engendrar un niño con DM1, generalmente más intensamente afecto que su madre. Se puede realizar un diagnóstico prenatal con una estimación aproximada de la afectación según el número de repeticiones de tripletes CTG.
Las mujeres con DM1 sufren con mayor frecuencia hidramnios, abortos espontáneos, debilidad durante el trabajo del parto, retención de placenta y hemorragia posparto117. Se ha descrito un 19% de partos pretérmino, un 13% de infecciones urinarias, placenta previa en un 9% y embarazos ectópicos en un 4% de los casos118. Por todo ello, además de las posibles complicaciones cardíacas de la gestante y las implicaciones anestésicas de la enfermedad, estas pacientes deben considerarse como embarazadas de alto riesgo, y controlarse obstétricamente con especial atención.
AnestesiaLos pacientes con DM1 tienen algunos riesgos específicos cuando son sometidos a procedimientos anestésicos, que deben tenerse en consideración. Es frecuente, en estos pacientes, intervenciones quirúrgicas para el tratamiento de las cataratas y cirugía abdominal, sobre todo la colecistectomía, además de las posibles intervenciones por otros trastornos21,119.
La anestesia locorregional es segura, pero los opiáceos y sedantes con efecto respiratorio debe utilizarse con cautela, puesto que estos pacientes son especialmente sensibles a estos fármacos117–120 y pueden, incluso, sufrir reacciones paradójicas con relajantes musculares. La anestesia raquídea es la más recomendable para la cirugía tocológica.
Para una anestesia general pueden usarse fármacos como propofol y opiáceos de acción corta, como remifentanilo119. En el período postoperatorio es conveniente vigilar la función respiratoria mediante pulsioximetría las primeras 24h, por el riesgo de apnea21,117,120 y de atelectasias por hipoventilación, en especial si la capacidad vital forzada está previamente alterada. Igualmente, existe un riesgo aumentado de arritmias cardíacas, lo que exige una monitorización del ritmo cardíaco120. No debe olvidarse el riesgo de una posible broncoaspiración.
En los pacientes que vayan a someterse a una anestesia general se recomienda siempre una evaluación respiratoria.
En los casos que siguen tratamiento con mexiletina no deben administrarse antiarrítmicos de clase i, por el riesgo de bloqueo de la conducción. Los relajantes musculares despolarizantes están contraindicados porque pueden inducir hiperpotasemia. Si son imprescindibles los relajantes musculares, se aconseja usar los que tiene una acción corta, recordando que pueden mostrar una acción más prolongada de lo normal. El uso de anticolinesterásicos, como prostigmina, puede producir intensa debilidad muscular.
La Deutschen Gesellschaft für Anästhesiologie und Intensivmedizin e. V. (Sociedad Alemana de Anestesiología y Medicina Intensiva) ha elaborado una guía de actuación anestésica para DM1 que puede consultarse traducida al español en el portal de Orphanet119.
Distrofia miotónica tipo 1 y cáncerSe ha descrito un aumento en la incidencia de tumores en los pacientes con DM1.
Las primeras neoplasias que se describieron en estos pacientes fueron los pilomatricomas, tumores cutáneos benignos, a menudo calcificados, derivados de células de la matriz del pelo. En diversos artículos se ha descrito la aparición de tumores de diferentes estirpes, incluyendo una posible asociación con carcinomas basocelulares múltiples. Un estudio que utilizó los registros sueco y danés de pacientes con distrofia miotónica entre 1977 y 2008121 encontró una incidencia de tumores mayor de la esperada. Las neoplasias de aparición aumentada con respecto a la población general fueron las de endometrio, cerebro, ovario y colon122. Este estudio no diferenciaba entre pacientes con DM1 y DM2, aunque dada la prevalencia relativa de ambas enfermedades es razonable pensar que la mayoría de los pacientes tuvieran una DM1. En una cohorte de 307 pacientes con DM1 y DM2 de la Clínica Mayo, recogida entre 1993 y 2010, se encontró un posible aumento en el riesgo de padecer cáncer de tiroides y melanoma coroideo, y quizás cáncer de próstata y testículos123. Otro estudio de base poblacional realizado en Utah encontró un exceso de tumores de endometrio, testículo, y linfomas no hodgkinianos124–126. Estos datos han sido validados en una cohorte española con una mayor prevalencia en mujeres siendo los tumores más prevalentes los de ovario y endometrio mientras que en hombres lo eran los de tiroides y cerebro127.
No se conoce la causa de este posible aumento de tumores en DM1, aunque se han apuntado diversas hipótesis que implican a la betacatenina o la regulación a la baja de una familia de miARN supresores (200c/141)127. Es interesante que no se haya encontrado aumento de incidencia de tumores en otras enfermedades por expansión de tripletes.
Se debe tener en cuenta el riesgo aumentado de tumores, ya que supone la tercera causa de muerte en estos pacientes. Las neoplasias con riesgo aumentado son en su mayoría tumores de baja incidencia, en los que no han demostrado clara utilidad los programas de cribado en la población general. Sí se recomienda la exploración de los pacientes en las consultas periódicas, con especial sospecha clínica ante síntomas sugestivos de cáncer, especialmente síntomas cerebrales o abdominopélvicos, o sangrado uterino. Se aconseja la palpación de tiroides en las revisiones endocrinológicas y estudio de imagen si se encuentran alteraciones. Se debe recordar la posibilidad de melanoma coroideo en la evaluación oftalmológica. La vigilancia es más importante en los tumores más frecuentes en la población general, como son los cutáneos.
Consideraciones especiales en la edad pediátricaLa DM1 se manifiesta habitualmente en la edad adulta, pero existen 2 formas de presentación pediátrica: la distrofia miotónica congénita y la variante de inicio infantil. Ambas tienen unas características diferenciales respecto a la forma clásica del adulto, aunque el proceso diagnóstico es similar: la sospecha clínica seguida de la confirmación genética mediante el estudio dirigido.
Distrofia miotónica congénitaEs la forma más grave, pero también la más infrecuente. La incidencia varía entre 2,1 y 5,2 por 100.000 recién nacidos vivos128,129. En España se ha estimado en 0,8 por 10.000130. En la gran mayoría de los casos la transmisión es materna, produciéndose una expansión intergeneracional masiva del número de tripletes. Es habitual que el niño con distrofia miotónica congénita sea el caso índice que permite diagnosticar a la madre, generalmente paucisintomática, y a otros miembros de la familia.
Las manifestaciones de la enfermedad están presentes desde el nacimiento, aunque los antecedentes obstétricos suelen reflejar la escasa movilidad del feto ya en el período prenatal: movimientos fetales reducidos, polihidramnios y deformidades articulares detectados mediante ecografía, o una gestación finalizada mediante cesárea debido a que el parto no progresa. Las dificultades en el expulsivo suponen un riesgo añadido de asfixia perinatal. El neonato muestra al nacer hipotonía marcada asociada a debilidad y pobreza de movimientos. La facies es característica con paresia facial y labio superior en forma de V invertida. Es muy común la presencia de problemas respiratorios (más del 50% de los casos) por debilidad del diafragma y de los músculos intercostales, inmadurez pulmonar (el riesgo de prematuridad está aumentado) y fallo del control respiratorio cerebral. También existen dificultades en la succión-deglución, debidas a la debilidad de la musculatura bulbar131. Las retracciones articulares son habituales, especialmente en forma de pie zambo o equino-varo. Al contrario que en las formas del adulto, los problemas cardiológicos no son frecuentes a esta edad y no existe miotonía clínica ni eléctrica hasta edades posteriores, por lo que no está indicada la realización de electromiograma. Se ha descrito una mayor incidencia de ventriculomegalia y de malformaciones del desarrollo cortical cerebral132.
El diagnóstico diferencial se establece preferentemente con las distrofias musculares y miopatías congénitas y con los síndromes miasteniformes congénitos.
La mortalidad neonatal de las formas congénitas se sitúa entre el 16 y el 41%128,133,134. Ello se debe esencialmente a problemas respiratorios y a la retirada de soporte en pacientes ventilados durante más de 4 semanas, en los que se asume una escasa posibilidad de recuperación a corto/medio plazo135. Sin embargo, este concepto está cambiando ante las evidencias histopatológicas que consideran la forma congénita como una entidad dismadurativa y, por lo tanto, con potencialidad de mejoría a lo largo del tiempo. En este sentido la duración de la ventilación no debería condicionar de forma aislada la continuidad de los cuidados médicos128.
Los niños supervivientes al sexto mes de vida mejoran lentamente, aunque manifiestan un retraso evidente en las habilidades motoras. Prácticamente todos consiguen la marcha autónoma, y hacia los 3-4 años mejora la hipotonía y las dificultades para la alimentación. La debilidad facial, en cambio, sigue siendo prominente ocasionando la típica «boca de carpa». La miotonía no aparece hasta la adolescencia. Frente a los progresos motores, en los primeros años se pone de manifiesto una afectación cognitiva de intensidad variable, aunque existe una minoría de casos con capacidad intelectual normal136. El cociente de inteligencia se sitúa de media en 70, pero el retraso mental puede llegar a un grado moderado o grave. Este es uno de los principales problemas de la forma congénita. Los niños pueden presentar rasgos del espectro autista y otros problemas conductuales asociados137. El aspecto facial puede condicionar que al niño se le atribuya una capacidad cognitiva menor de la que realmente tiene. El desarrollo del lenguaje puede estar interferido tanto por la debilidad en la musculatura de la boca, paladar y mandíbula, que dificultan una buena pronunciación, como por el riesgo de hipoacusia ante las infecciones repetidas del tracto respiratorio superior. Con los años aparece la debilidad en extremidades con distribución distal, así como el resto de las manifestaciones propias de la edad adulta.
Distrofia miotónica de inicio infantilEn la forma infantil las manifestaciones clínicas aparecen entre el primer y el décimo año de vida, siendo normal el desarrollo pre, peri y posnatal normal durante los primeros 12 meses. Esta variante clínica tiene poco que ver con la forma congénita. La transmisión puede ser materna o paterna. Las manifestaciones motoras pueden no ser llamativas inicialmente y destacan los problemas del lenguaje y las dificultades escolares. En este contexto se debe tener un índice alto de sospecha para poder diagnosticar la enfermedad a esta edad138. Hay trastornos conductuales, el déficit de atención e hiperactividad es el problema más frecuente, y es posible la afectación visuoespacial incluso con un cociente de inteligencia normal29. Existe correlación entre la afectación cognitiva y el tamaño de la expansión de tripletes139.
En los casos con afectación muscular inicial puede haber torpeza en los movimientos y debilidad en la musculatura facial y del cuello, pero sin el aspecto típico de las formas congénitas134. También es posible encontrar debilidad en extremidades con distribución distal y miotonía clínica, como ocurre en edades posteriores. En ocasiones las molestias gastrointestinales son persistentes por implicación de la musculatura lisa. Es rara la cardiopatía, que aparece a partir de la segunda década de la vida. Tampoco se detectan cataratas, características en la edad adulta, pero hay mayor frecuencia de otros problemas oftalmológicos como defectos refractivos, reducción de agudeza visual y alteraciones oculomotoras139,140. Con el tiempo aparecen las mismas complicaciones que en las formas del adulto.
Manejo de la distrofia miotónica tipo 1 en la edad pediátricaEl tratamiento debe ser multidisciplinar y abarcar todos los problemas de la enfermedad131. Los nacidos con distrofia miotónica congénita deben ser atendidos en una unidad neonatal de cuidados intensivos, vigilando especialmente la función respiratoria. Según la situación se planteará la necesidad de ventilación asistida, no invasiva o mediante traqueostomía. La decisión de limitar el esfuerzo terapéutico debe ser individualizada, pues, como ya se ha comentado, la necesidad de ventilación prolongada no conlleva obligatoriamente un mal pronóstico a medio/largo plazo. En los supervivientes se debe mantener el seguimiento neumológico a lo largo de la infancia. La evaluación respiratoria también en necesaria en las formas de inicio infantil por la posible aparición de apnea obstructiva del sueño y somnolencia diurna. Los problemas de succión-deglución deben ser estudiados para evitar las aspiraciones, y se planteará la necesidad de alimentación por sonda o gastrostomía. Las alteraciones cognitivas y conductuales son especialmente relevantes en la infancia, por lo que se debe realizar una evaluación psicopedagógica completa que incluya la evaluación de la capacidad cognitiva, así como de las funciones ejecutivas y visuoespaciales. Es necesaria la coordinación con el centro escolar para realizar una adecuada intervención psicoeducativa.
Las complicaciones cardiológicas son raras antes de la segunda década de la vida, pero existe la posibilidad de cardiomiopatía y alteraciones de la conducción cardíaca, por lo que se recomiendan revisiones cardiológicas periódicas con ECG anuales141,142.
Desde el punto de vista motor el niño debe actuar en función de sus capacidades y no se debe restringir su actividad física131. Puede beneficiarse de terapia ocupacional y de programas de fisioterapia dirigidos. Los problemas orofaciales deben ser abordados por logopedas. El tratamiento ortopédico, mediante ortesis o cirugía, está indicado si existen deformidades articulares143. Es recomendable realizar evaluaciones oftalmológicas y auditivas para prevenir tanto la ambliopía como la hipoacusia.
En los procedimientos anestésicos debe extremarse la precaución ante la posibilidad de disritmias cardíacas y apneas. Se debe mantener una observación estrecha en las primeras horas tras la intervención.
El consejo genético cobra especial importancia en los casos pediátricos al tratarse de familias con progenitores jóvenes que pueden querer aumentar la descendencia.
Documentos para atención en urgencias y emergencias de la distrofia miotónica tipo 1Cuando un paciente con DM1 acude a un servicio de urgencias puede producirse una cierta incertidumbre que dificulte el manejo de cualquier problema médico, sea uno propio de la enfermedad, sea un problema común. Para asistir a los médicos de urgencias y a los pacientes se idearon las tarjetas de urgencias para facilitar el acceso rápido a los datos médicos relevantes de los pacientes y a guías de actuación de urgencia con información clínica contrastada. Se recomienda facilitar el acceso a esta información a los pacientes, familiares y cuidadores y fomentar el uso de tarjetas de urgencia.
La Société Française de Médecine d’Urgence (SFMU) realizó una encuesta para conocer las expectativas de los médicos de urgencias respecto a los pacientes afectados por enfermedades minoritarias144. El 91% de los encuestados deseaban que los enfermos llevasen una tarjeta con sus datos personales y recomendaciones para el tratamiento de su enfermedad en urgencias. Aproximadamente el 75% de los facultativos deseaban contar con un servicio en línea (vía Internet) que pusiese a su disposición una guía práctica para estas situaciones.
En respuesta a estas necesidades, se crearon en Francia nuevos servicios dirigidos a los profesionales de la salud, con el fin de mejorar la información sobre enfermedades raras que debe estar disponible en Internet mediante la implementación del programa Orphanet Urgences («Buenas prácticas en casos de urgencias»), desarrollado con el Service d’Aide Médicale Urgente y los profesionales de los servicios de urgencias hospitalarias. Estos servicios son guías de actuación de urgencia y tarjetas de cuidados y urgencias para algunas enfermedades, la DM1 entre ellas, de acceso libre y gratuito a través de Internet57,145. Su grado de utilización, cuantificado a través del número de descargas, es satisfactorio146.
No existen investigaciones similares en nuestro medio, pero es razonable extrapolar los resultados de esta encuesta al contexto asistencial de España. Se ha pretendido responder a las 2 necesidades expresadas por los facultativos de urgencias (información aportada por el paciente e información en línea) confeccionando para los enfermos con DM1 una denominada «Tarjeta de Alerta Médica». Se trata de un documento de bolsillo que el enfermo debe llevar siempre consigo, destinado a los profesionales de urgencias normalmente no familiarizados con la enfermedad. Contiene una breve descripción de la dolencia, datos de contacto del profesional de referencia, un enlace QR a la guía de urgencias de www.orpha.net/ para la DM1 y puede alojar información actualizada sobre la evolución del paciente147.
Cabe destacar que varias entidades vienen elaborando y difundiendo documentos de urgencias para los afectados por DM1 (las asociaciones británicas Muscular Dystrophy UK y Myotonic Dystrophy Support Group, la Scottish Muscle Network e, incluso, centros hospitalarios del National Health Service, también en el Reino Unido).
Pese al riesgo de extravío, deterioro u olvido del documento, a la limitada información que puede contener, y al margen de un posible efecto tranquilizador para el paciente, la «Tarjeta de Alerta Médica» para DM1 es principalmente un medio para facilitar, mediante las nuevas tecnologías, un acceso rápido a información validada por expertos. Lamentablemente, todavía no ha sido posible cuantificar su grado de utilización148.
FinanciaciónEste artículo no ha recibido financiación para su elaboración.
Conflicto de interesesNinguno.
Esta Guía de Consenso ha obtenido los avales científicos de la Sociedad Española de Neurología (SEN), de la Sociedad Española de Endocrinología y Nutrición (SEEN) y Grupo de Cardiopatías familiares de la Sociedad Española de Cardiología (SEC).
Artículo publicado previamente en: Medicina Clínica: 10.1016/j.medcli.2018.10.028, con el consentimiento de los autores y editores.
recomendados
Neurology Perspectives


Neurología sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas