



La exposición de líquido cefalorraquídeo (LCR) de pacientes con esclerosis lateral amiotrófica (ELA) induce efectos citotóxicos en cultivos celulares de neuronas motoras in vitro.
Material y métodosSe seleccionó LCR de 32 pacientes con ELA que previamente habían demostrado efectos citotóxicos. Se implantaron con minibombas osmóticas intracerebroventriculares (ICV) en 28 ratas macho adultas y se dividieron en 3 grupos: 9 ratas de LCR de pacientes no-ELA, 15 ratas de ELA-LCR citotóxico y 4 ratas de una solución salina fisiológica. El LCR se administró por vía ICV de forma continua durante periodos de 20 o 43días. Se realizó la evaluación clínica, electromiográfica y análisis de tejidos después de sacrificio a los 20, 45 y 82días tras la cirugía.
ResultadosLos estudios inmunohistoquímicos muestran daño en los tejidos con características similares a las encontradas en formas esporádicas de ELA, tales como sobre expresión de cistatinaC, transferrina y la proteína en el TDP-43 citoplasmática. Los primeros cambios observados parecían jugar un papel protector por la sobreexpresión de periferina, panAKT, fosfoAKT y metalotioneínas; esta expresión habría disminuido al momento de analizar las ratas que se sacrificaron al día 82, en el que hay un aumento de apoptosis. Los primeros cambios celulares identificados fueron la constatación de activación de la microglía seguido por astrogliosis con sobreexpresión de GFAP y proteína S100B.
ConclusionesNuestros datos parecen indicar que la ELA podría propagarse a través del LCR, y que la administración ICV de ELA-LCR citotóxico produce cambios similares a los encontrados en las formas esporádicas de la enfermedad.
Cerebrospinal fluid (CSF) from amyotrophic lateral sclerosis (ALS) patients induces cytotoxic effects in in vitro cultured motor neurons.
Material and methodsWe selected CSF with previously reported cytotoxic effects from 32 ALS patients. Twenty-eight adult male rats were intracerebroventricularly implanted with osmotic mini-pumps and divided into 3 groups: 9 rats injected with CSF from non-ALS patients, 15 rats injected with cytotoxic ALS-CSF, and 4 rats injected with a physiological saline solution. CSF was intracerebroventricularly and continuously infused for periods of 20 or 43days after implantation. We conducted clinical assessments and electromyographic examinations, and histological analyses were conducted in rats euthanised 20, 45, and 82days after surgery.
ResultsImmunohistochemical studies revealed tissue damage with similar characteristics to those found in the sporadic forms of ALS, such as overexpression of cystatinC, transferrin, and TDP-43 protein in the cytoplasm. The earliest changes observed seemed to play a protective role due to the overexpression of peripherin, AKTpan, AKTphospho, and metallothioneins; this expression had diminished by the time we analysed rats euthanised on day 82, when an increase in apoptosis was observed. The first cellular changes identified were activated microglia followed by astrogliosis and overexpression of GFAP and S100B proteins.
ConclusionOur data suggest that ALS could spread through CSF and that intracerebroventricular administration of cytotoxic ALS-CSF provokes changes similar to those found in sporadic forms of the disease.
La esclerosis lateral amiotrófica (ELA) tiene como característica principal la pérdida selectiva de neuronas motoras en el cerebro, el tronco cerebral y la médula espinal1. La mayoría de los casos de ELA son esporádicos, pero aproximadamente el 10% de los pacientes tienen un historial familiar2, y de estos pacientes, el 20% tienen mutaciones en el gen de Cu2+/Zn2+ superóxido dismutasa1 (SOD1)3. Además, la TDP-43 y la proteína FUS/TLS podrían estar asociadas en pacientes con ELA familiar y esporádica4,5, y el mayor porcentaje de casos han sido vinculados a una expansión intrónica de una repetición GGGGCC del hexanucleótido en el gen C9ORF726.
El mecanismo por el que las neuronas motoras se convierten en selectivamente vulnerables y mueren en pacientes con ELA es aún desconocido. El patrón de progresión clínica encontrado en ratones transgénicos mutados de SOD1 podría indicar que la enfermedad se propaga de forma centrífuga desde una región afectada a una zona adyacente; por lo tanto, se podría especular que productos tóxicos generados en un área difundan la lesión, afectado a las neuronas adyacentes y de este modo propagando la enfermedad, y una posibilidad es que estos factores podrían ser vehiculados a través del líquido cefalorraquídeo (LCR)7. Esta suposición llevó a varios investigadores a desarrollar experimentos para investigar si la incubación in vitro en cultivos de neuronas y de líneas de células neuronales con el LCR de pacientes con ELA (ELA-LCR) podría causar daño celular, lo que se confirmó en la mayoría de los estudios7-22 como revisión, aunque no en todos23. Nuestro grupo ha determinado que ELA-LCR afecta la viabilidad de neuronas motoras corticales de cerebro de rata mantenidas en cultivos primarios; sin embargo, este efecto parece que está mediado por un mecanismo independiente de glutamato, aunque la ELA-LCR causó una elevación de la concentración basal de Ca2+ citosólico ([Ca2+]) de las neuronas motoras24,25. Estos datos apoyan la presencia de factor(es) tóxico(s) en la ELA-LCR responsable(s) de provocar daño de las neuronas motoras en experimentos in vitro, pero es posible que no en todos los pacientes o en todas las fases de la enfermedad26. La pregunta es si la ELA-LCR también podría causar daño cerebral in vivo. De hecho, la inyección intratecal o intracerebroventricular (ICV) aguda de ELA-LCR produce cambios en la actividad neuronal, en la expresión de los receptores de glutamato y canales iónicos, en la fosforilación de proteínas al cabo de solo 2días después de la inyección27-29 o induce alteraciones mitocondriales, estrés oxidativo o disfunción lisosomal30.
Estos experimentos producidos con inyecciones puntuales no imitan el curso temporal de la ELA y por lo tanto no la reproducen. Por lo tanto, parecía apropiado llevar a cabo los experimentos que describiremos aquí: el examen de los cambios cerebrales histopatológicos y funcionales que se producen después de la infusión continua de ELA-LCR durante 20 o 43días, logrado mediante el uso de una minibomba conectada al ventrículo cerebral de rata a través de una cánula, a fin para determinar las consecuencias de la exposición más prolongada de ELA-LCR citotóxico. Aunque no encontramos cambios funcionales similares a la ELA, encontramos mayor expresión temprana de moléculas de señalización de neuroprotectores, una reacción neuroinflamatoria temprana acompañada de microglía activada y astrogliosis, así como la sobreexpresión de las proteínas citoplasmáticas ELA-vinculadas como TDP-43, cistatinaC y transferrina.
Material y métodosRecolección de líquido cefalorraquídeo y preparación de ELA-LCR citotóxicos (cELA-LCR)Las muestras de LCR fueron recolectadas por punción lumbar en 32 pacientes con ELA diagnosticados según los criterios de diagnóstico de El Escorial31 (25).** Fueron extraídos entre 1,5 y 3cc de cada paciente, centrifugados a 1.500rpm durante 10min, y divididos en 4 a 5 alícuotas para propósitos del estudio. Se obtuvo un consentimiento informado de cada paciente. Dieciséis pacientes eran mujeres; la edad media de la serie fue de 59años al momento de la extracción del LCR. Cuatro pacientes tenían ELA familiar y el resto eran esporádicos; 11casos fueron presentados como una forma de aparición bulbar, 20 casos sufrieron el tipo de la columna vertebral, y un paciente presenta síntomas en ambos lugares. De los pacientes con ELA, solo 4 presentaban mutaciones en el gen SOD1 y uno era asintomático, lo que significa que el LCR de pacientes con un mecanismo de genética SOD1 tenía una muy baja presencia en el grupo ELA-LCR. Veinte muestras (68,7%) mostraron una citotoxicidad significativa (cELA-LCR) usando el método descrito a continuación. Las muestras de LCR utilizadas como controles (no-ELA LCR) fueron obtenidas de pacientes que sufren dolor de cabeza o convulsiones y requirieron punción lumbar durante un examen de rutina. Se obtuvo también un consentimiento informado de los controles. Las muestras de LCR se mantuvieron congeladas a −80°C hasta que fueron utilizadas.
Con la finalidad de demostrar citotoxicidad, fueron utilizados en los experimentos cultivos celulares de neuronas motoras in vitro, como ha sido descrito previamente24. Para estos experimentos, las neuronas se obtuvieron de la corteza motora cerebral de embriones de rata de 20días de edad. Las células obtenidas se sembraron a una densidad de 105células/ml en placas de 48 pocillos que contenían 0,25ml de medio neurobasal suplementado con B-27 y cultivado durante 8-10días. Después de este tiempo, los cultivos se incubaron durante 24h con 10% ELA-LCR y la viabilidad de las neuronas fue medida con ensayos MTT32. La ELA-LCR administrada a los animales fue preparada por la puesta en común de viales de LCR que exhibían efectos citotóxicos in vitro (por lo menos 20% de pérdida de las neuronas motoras), y la mayoría del LCR incluido en ese grupo fue tomado de pacientes con ELA esporádica. Las muestras de LCR control para los no-tóxicos de 3 pacientes también fueron considerados antes de la administración ICV.
AnimalesLos animales fueron manipulados según los principios de cuidado de animales de laboratorio publicados por el Comité Ético Español (RD 1201/2005) y la Directiva Europea 86/609/CEE; el procedimiento fue aprobado por el Comité de Ética para el cuidado y uso de animales de investigación en el Hospital Clínico San Carlos de Madrid, España. Las 28 ratas Wistar albinas macho adultos fueron obtenidas de Charles River Laboratories. Todos los animales fueron alojados bajo un ciclo de luz/oscuridad de 12h con acceso libre a comida y agua. Fueron enjaulados individualmente bajo condiciones de colonias estándar en las instalaciones de los animales Hospital Clínico San Carlos. El grupo LCR se administró a las ratas en 4 períodos. En el primer período, 14 ratas entre 1 y 5meses de edad (media de 3,25meses) y en el segundo periodo, 12 ratas entre 2,5 y 5meses de edad (media de 4,1meses) fueron inyectadas con el LCR descrito anteriormente. El intervalo entre los 4 períodos fue de aproximadamente 3meses.
Grupos experimentalesTres grupos experimentales fueron diseñados para este estudio. El grupo1 incluyó 9 ratas a las que fueron implantadas ICV minibombas llenas de LCR de pacientes no-ELA (no-ELA-LCR). El grupo2 fue compuesto por 15 ratas implantadas ICV con minibombas llenas de ELA-LCR citotóxico (cELA-LCR). Finalmente, 4 ratas implantadas ICV con minibombas llenas de solución salina fisiológica formaron el grupo3 (grupo de tratamiento simulado o sham).
Administración intracerebroventricular de cELA-LCR o no-ELA-LCRLas ratas fueron anestesiadas con tribromoetanol, y se les implantó por vía subcutánea una minibomba osmótica (caudal de 0,15l/h; Alzet 2006, Alzet Palo Alto, CA, EE.UU.) entre las escápulas y permaneció allí hasta el punto final del estudio. Antes del implante, las minibombas osmóticas se llenaron con la preparación correspondiente para cada grupo experimental. Las minibombas se adjuntaron a una cánula de infusión de cerebro (Alzet) a través de un tubo de polietileno y son preparadas por inmersión en solución salina normal a 37°C durante 24h. Estos procedimientos fueron llevados a cabo en condiciones estériles. La cánula fue implantada en el ventrículo cerebral lateral derecho (−0,5mm anteroposterior, −1,4mm lateral y −3,3mm dorsoventral (coordenadas basadas en el Atlas Paxinos)33 y fueron fijadas al cráneo con cemento dental.
El volumen de depósito de las bombas asegura el bombeo al menos durante 42días. El LCR fue inyectado por vía intratecal a una tasa de 0,15l/h durante un período de 43días, excepto las ratas sacrificadas al día 20. Los animales restantes en el estudio que permanecieron hasta el día 82 fueron implantadas con un tubo de polietileno sellado mecánicamente para prevenir la irritación local debido a la continua atracción de agua a la bomba.
Evaluaciones clínicasLos animales fueron probados clínicamente al menos un día por semana a partir de una semana después de la cirugía. Todas las pruebas se realizaron con los examinadores ciegos al grupo experimental al que pertenecía cada rata. Las evaluaciones del estudio incluyeron la medición de peso corporal, la respuesta a la prueba de plano inclinado y la escala motora Matsumoto34. Para medir el peso corporal, los animales se pesaron cada 4semanas con una escala digital. Para la prueba de plano inclinado, 20 ratas fueron colocadas lateralmente contra el eje largo del plano inclinado para medir el ángulo máximo en el que podían mantener su posición en el plano durante 5s. Para evaluar la fuerza de ambas extremidades por igual, los animales fueron colocados sobre el plano inclinado con su lado derecho del cuerpo frente a la parte descendente de la superficie inclinada, posteriormente con el lado izquierdo del cuerpo que va cuesta abajo. Una puntuación de menos de 70¿ en la prueba de plano inclinado se correlaciona bien con la aparición de debilidad muscular en ratones transgénicos SOD1-G93A34. Para determinar la escala motora de Matsumoto34 se puso a prueba la capacidad de los animales de enderezarse a sí mismos dentro de los 30s de ser manipulado a ambos lados (reflejo de enderezamiento). Las ratas que eran capaces de enderezar desde ambos lados fueron examinadas para su capacidad de permanecer en su jaula de alojamiento de forma espontánea durante 1min. Si mostraban poco movimiento en sus jaulas, se trasladaban a otra jaula para activar la motivación de exploración. Si las ratas no mostraron anomalías de acuerdo con estas evaluaciones, eran examinadas a fondo para ver si tenían algún déficit funcional observable, tal como parálisis de las extremidades o síntomas de debilidad muscular general en el campo abierto. Una puntuación de 5 significa que el animal es normal y una puntuación de 0 es una rata completamente paralizada. La escala motora de Matsumoto ha demostrado estar correlacionada con la pérdida de neuronas motoras espinales en un modelo de ratón transgénico mutado de ELA SOD1-G93A34
Examen electromiográficoUn electromiograma de las ratas cELA-LCR y ratas no-ELA-LCR fue realizado al día 82 por un neurólogo experto en esta técnica. La aguja fue insertada en las extremidades delanteras y traseras y en los músculos paravertebrales para tratar de hallar signos de denervación como actividad espontánea de inserción (ondas positivas, fibrilaciones o fasciculaciones). Las ratas fueron sedadas con isoflurano al 1,5% en 0,7l/min de oxígeno durante el procedimiento.
Sacrificio de los animalesA los 20, 45 u 82días después de la cirugía las ratas de cada grupo fueron profundamente anestesiadas con una dosis de pentobarbital (60mg/kg) y fentanilo (0,3mg/kg). En primer lugar, se inyectó 10μl de colorante de Evans azul a través de un catéter ventricular para confirmar la correcta localización. Los animales fueron sacrificados por perfusión intracadiaca con 0,9% de solución salina seguido de paraformaldehído al 4% tamponado (tampón fosfato 0,1M). Después de la perfusión, el encéfalo y la médula espinal fueron diseccionados, lavados con PB 0,1M y crioprotegidos por inmersión en 30% de sacarosa y compuesto OCT. El material se almacenó a −80°C hasta su uso. Se cortaron criosecciones (50μm) de la corteza motora y de los segmentos cervical y lumbar de la médula espinal (C5-C6 y L3-L5); los cortes fueron colocados en una solución de crioprotección que contiene etilenglicol y sulfóxido de dimetilo y es utilizado para preservación de tejidos cerebrales. De las 9 ratas en el grupo de no-ELA-LCR, una fue sacrificado al día 20, 7 a los 45días y una al día 82. De las 15 ratas en el grupo de cELA-LCR, 4 fueron sacrificados al día 20, 7 al día 45 y 6 al día 82 después del implante. Finalmente, de las 4 ratas en el grupo de tratamiento simulado, una fue sacrificada al día 20, 2 a los 45días y una a los 82días.
Análisis de histoquímica e inmunofluorescenciaSe lavaron 4 cortes de tinción de inmunofluorescencia con PBS, permeabilizados con 0,1% Triton X-100 y bloqueados con 10% de suero de cabra normal. Después se aplicaron los siguientes anticuerpos primarios durante la noche a 4°C: anti-periferina (1:200, Millipore, AB 9282); anti-S100B (1:200, Millipore, 04-1054); anti-caspasa-3 (1:200, Millipore, 04-1090); anti-proteína ácida fibrilar glial (GFAP) (1:500, DakoCytomation, Z0334); anti-GTL1 (1:200, Millipore, AB 1783); anti-GFAP (1:600, Millipore, MAB360); anti-TARDBP (1:200, Abcam, ab42474); anti-ubiquitina (1:100, Abcam ab7780); anti-transferrina-r (1:200, Abcam, ab22391); anti-colin acetiltransferasa (1:100, Abcam, ab68779); anti-metalotioneína (1:100, Abcam, ab12228); anti-Stefin (1:100, Abcam, ab68290); anti-MHC II (1:500, Abcam, ab6403); anti-Iba-I (1:1.000, Wako, desde 019-19741); anti panAKT (cell signaling 2920S) y anti fosfoAKT (cell signaling 4060S). Se lavaron los portaobjetos en PBS 3veces, y los anticuerpos primarios fueron probados con Cy3 (1:1.000, Jackson) o con anticuerpos secundarios conjugados Alexa 488, 555 o 647 (1:500, Invitrogen). Se lavaron las secciones 3veces en PBS, contrastadas con DAPI, y montadas con el reactivo Fluorsave (Calbiochem).
Cuantificación de datos inmunohistoquímicosLas imágenes de fluorescencia fueron adquiridas con un microscopio confocal Olympus AF1000. Se llevó a cabo un estudio inmunohistoquímico descriptivo dentro de los 3 períodos de tiempo establecidos, un análisis de los cambios observados en los marcadores inmunohistoquímicos con el tiempo y su comparación entre los grupos control y ELA-LCR. Los análisis histológicos examinaron las áreas estrechamente relacionados con ELA, específicamente la corteza motora, bulbo raquídeo y segmentos espinales C5-C6 y L3-L5.
GFAP, IBA1, caspasa-3 y MHC-IILas imágenes confocales fueron adquiridas con la misma configuración y distribución de fluorescencia utilizando Olympus Confocal Software (Olympus FluoView FV1000). Se realizó el análisis cuantitativo utilizando la versión 1.42q del software de análisis ImageJ (EE.UU.). El número de células positivas de inmunohistoquímica fue contado tomando al azar 368μm2 de cada área analizada de 3 a 4 diferentes secciones del cerebro coronal.
Periferina, S100B, panAKT, fosfoAKT y medición de metalotioneínaPara medir S100B, panAKT, fosfoAKT, metalotioneína y periferina, se siguió una versión modificada del método físico disector descrito por Reed, utilizando el principio de ladrillo imparcial del disector 3D35. El análisis fue realizado mediante mediciones de densidad óptica (procesamiento de imagen binaria de blanco y negro), obteniendo el porcentaje de área del campo analizado. Se seleccionaron 8 áreas por animal al azar por un experimentador ciego utilizando el software ImageJ 1.42 (NIH).
Análisis estadísticoLos datos fueron procesados utilizando el software GraphPad Prism 5. Fueron reportados como valores medios ±error estándar de la media (SEM). Los gráficos de los resultados fueron realizados utilizando el mismo programa. El criterio para la significación estadística fue p<0,05.
ResultadosTodos los animales sobrevivieron al implante ICV de la minibomba osmótica y ninguno de ellos mostró signos de infección (absceso, edema o secreción purulenta en el implante); ninguno sufrió convulsiones, agresión, piloerección o postración. La infusión fue realizada durante 20 o 43días y el curso temporal de los efectos de LCR o infusión de solución salina se evaluó a los 20, 45 y 82días después del implante de la bomba miniosmótica.
Evaluaciones clínicas y electromiográficasNo se encontraron diferencias significativas entre las ratas del grupo de cELA-LCR y los demás grupos en ninguna de las evaluaciones clínicas, incluyendo el peso corporal, el comportamiento durante la prueba de plano inclinado y las escalas de Matsumoto, que fueron todas normales. Respecto a los electromiogramas, se encontraron ondas agudas positivas en las extremidades traseras, las extremidades delanteras y los músculos paravertebrales sin potenciales de fibrilación o fasciculación en 6 de las ratas del grupo cELA-LCR a los 82días después del implante, lo que no ocurrió en los controles, aunque no se observó un patrón de denervación.
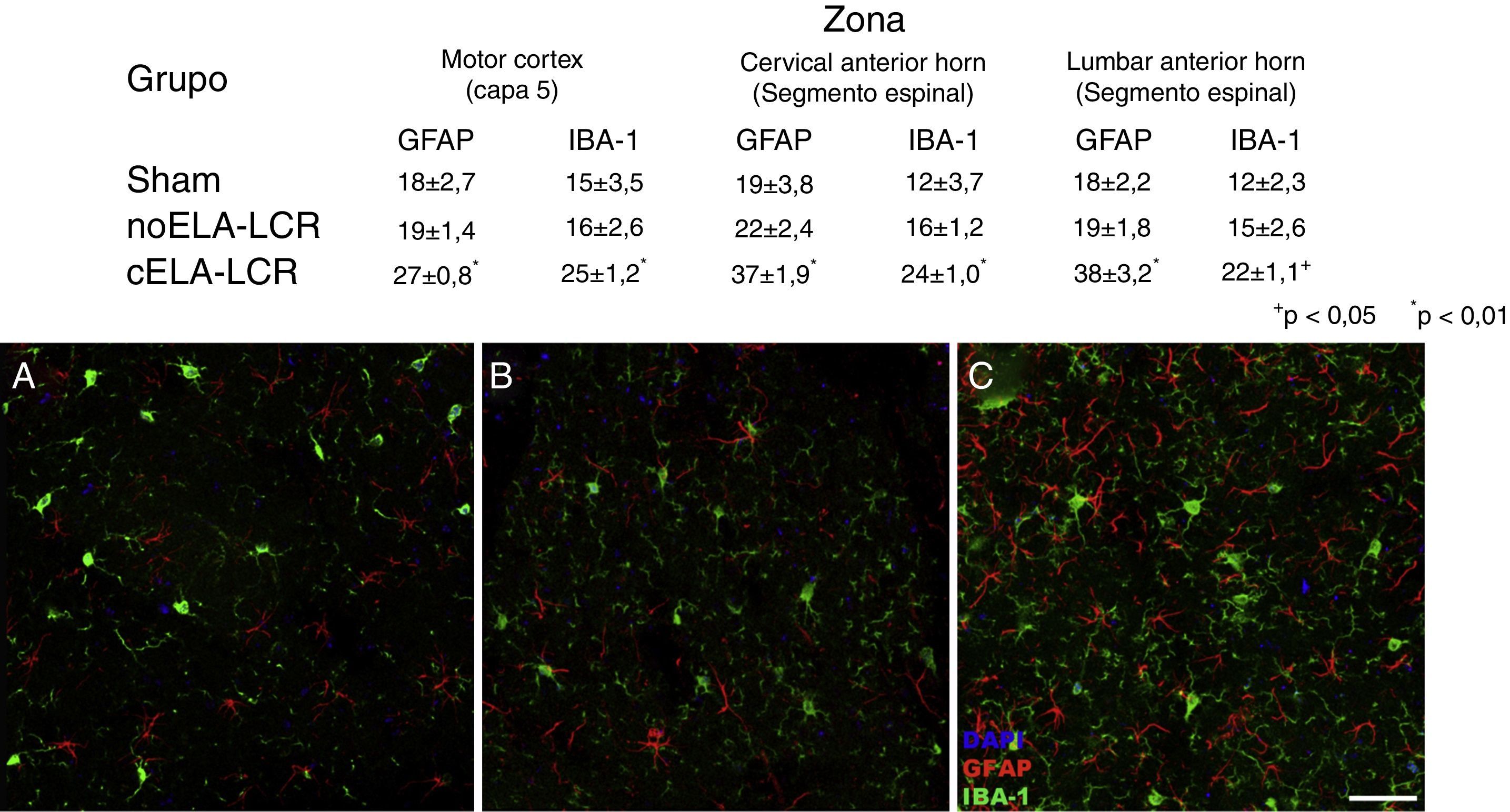
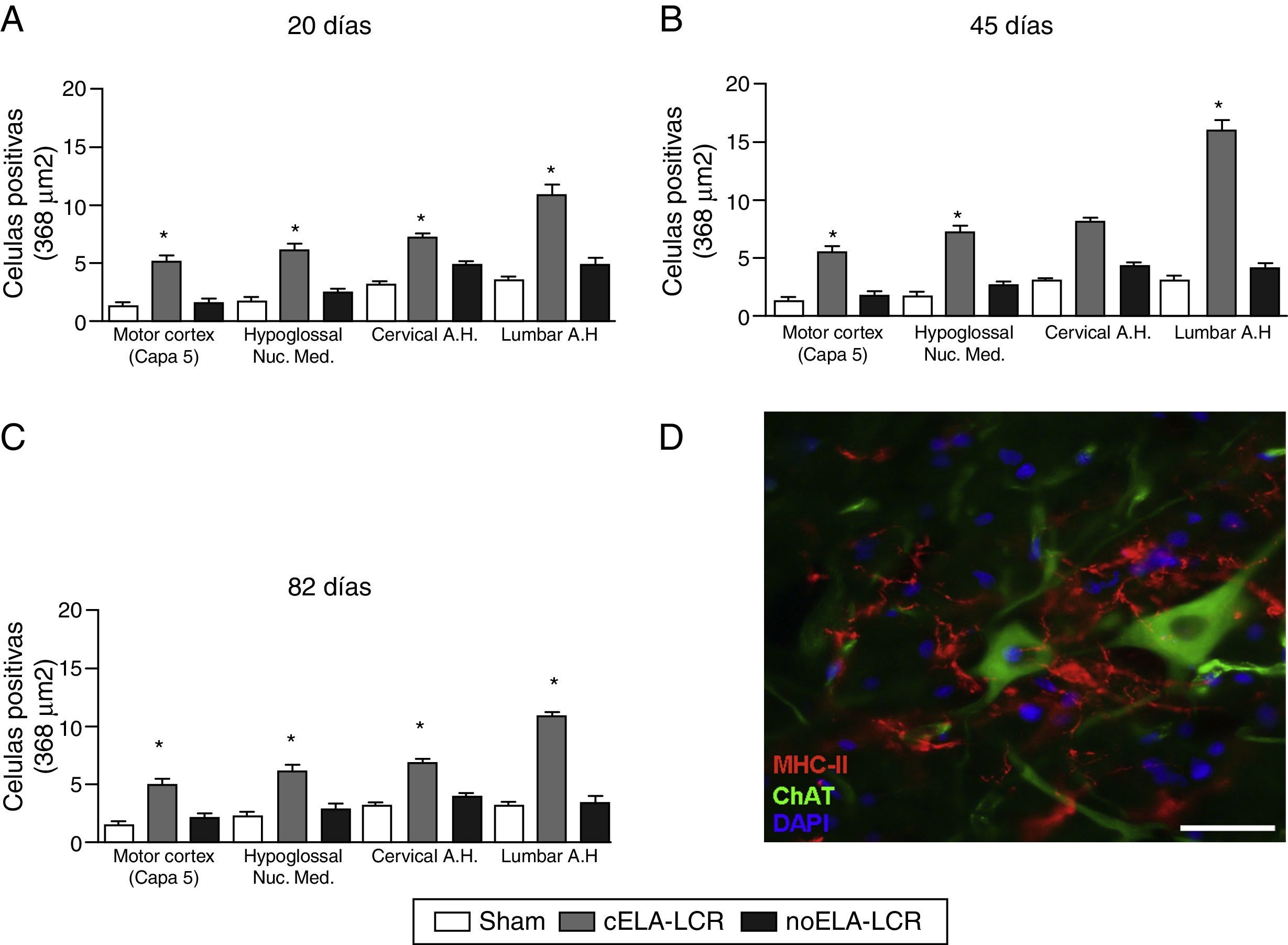
Activación microglial y expresión de GFAP, S100B, GLT1 y la caspasa-3Se observó gliosis reactiva evidente, activación microglial y expresión de caspasa-3 en la corteza motora en los 3 grupos experimentales a los 20días después del implante. Estos hallazgos podrían ser atribuidos a una respuesta inflamatoria secundaria a la intervención quirúrgica. Sin embargo, a los 45días después de la cirugía se observó un importante incremento en la reactividad glial y la respuesta microglial en todas las regiones en el grupo de cELA-LCR (figs. 1 y 2). En los segmentos de la médula espinal de los animales que recibieron cELA-LCR se observó que algunas motoneuronas estaban envueltas por células microgliales. La cuantificación inmunohistoquímica del marcador IBA-1 reveló alteraciones en la morfología de la microglía, pasando de un estado en reposo caracterizado por un soma microglial con procesos finos y ramificados a una microglía activada con escasas ramificaciones. Se observaron cuerpos celulares alargados con procesos largos y más gruesos a los 20días después del implante, mientras que se encontraron las células ameboides con cuerpos redondos con procesos cortos, gruesos y robustos a los 45 y 82días, lo que indica una activación de células fagocíticas en la proximidad de las neuronas motoras. Estas células expresan MHC-II, que marca fases inflamatorias. En los grupos de tratamiento simulado y no-ELA-LCR, la expresión de MHC-II fue 3 veces inferior a la observada en el grupo de cELA-LCR (fig. 3). Un punto interesante fue la fuerte expresión de astrocitos y el descubrimiento de astrocitos hipertróficos que sobreexpresaban GFAP en las proximidades de las neuronas motoras corticales y de la médula espinal. Se observaron cambios más pronunciados a los 45días después de la cirugía. También se observó expresión de MHC-II, pero a niveles más bajos, a los 82días (fig. 3). Contrariamente a los datos observados en el grupo de cELA-LCR, se constató una disminución de la expresión de GFAP y de caspasa-3 en los otros 4 grupos, incluso a los 82días después de la cirugía. Asimismo, se observó un ligero aumento en la expresión del transportador de glutamato GTL1, que se ha relacionado con la ELA, y su expresión está modificada por ELA-LCR36. GTL1 se encontró en las proximidades de las neuronas motoras espinales del grupo cELA-LCR que se correlaciona con la expresión de GFAP como un marcador de activación de astrocitos. La sobreexpresión de S100B apareció a los 45 y 82días en el grupo de cELA-LCR solamente.
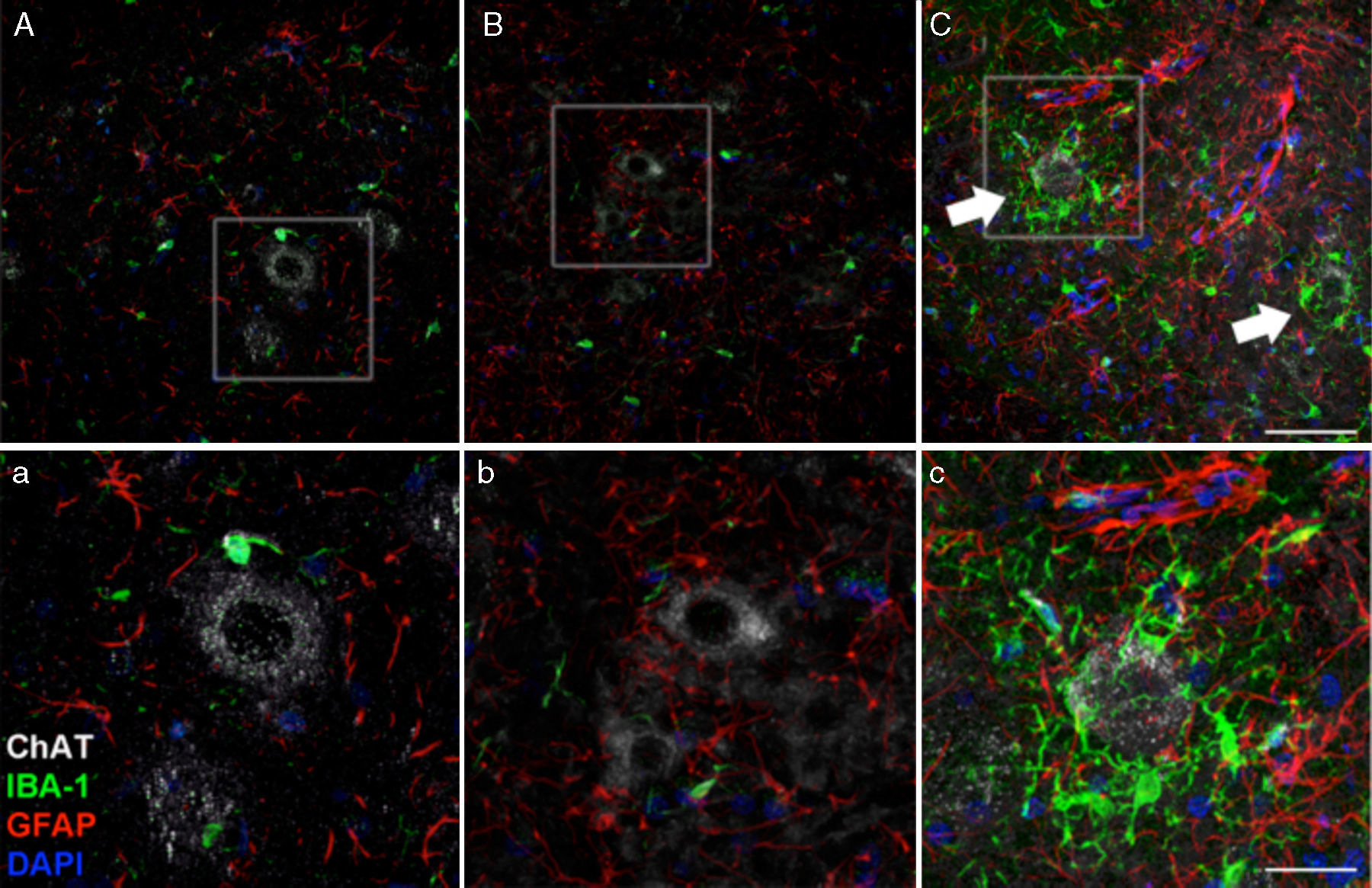
 de ratas cELA-LCR, encontramos una mayor expresión de GFAP con abundantes astrocitos hipertróficos (panel C, c); además, se identificó una mayor densidad de células IBA-1 positivas con procesos agrandados, a menudo en estrecha proximidad a las neuronas motoras o incluso rodeando a estas neuronas (flechas en panel C, flechas y detalle del panel c). Estos cambios no se observaron en los grupos sham y no ELA-LCR, en los que las células de microglía aisladas se observaron ocasionalmente cerca de las neuronas motoras. Escala: A-C: 60μm; detalles a-c: 30μm.")
A los 45 días después de la cirugía, el aumento de la reactividad glial y la respuesta microglial en el grupo de cELA-LCR en todas las regiones cerebrales estudiadas fue evidente. Por ejemplo, en el cuerno anterior (segmento lumbar) de ratas cELA-LCR, encontramos una mayor expresión de GFAP con abundantes astrocitos hipertróficos (panel C, c); además, se identificó una mayor densidad de células IBA-1 positivas con procesos agrandados, a menudo en estrecha proximidad a las neuronas motoras o incluso rodeando a estas neuronas (flechas en panel C, flechas y detalle del panel c). Estos cambios no se observaron en los grupos sham y no ELA-LCR, en los que las células de microglía aisladas se observaron ocasionalmente cerca de las neuronas motoras. Escala: A-C: 60μm; detalles a-c: 30μm.
. Es evidente el aumento de células positivas IBA-1 y GFAP en los animales del grupo cELA-LCR (C), en comparación con el grupo sham (A) y el grupo de noELA-LCR (B). Corteza motora capa 4-6, 45días después de la cirugía. Escala de barras: 50μm.")
Imagen de microscopia confocal de la corteza motora, ejemplificando los datos presentados en tablas (diferentes momentos del estudio). Es evidente el aumento de células positivas IBA-1 y GFAP en los animales del grupo cELA-LCR (C), en comparación con el grupo sham (A) y el grupo de noELA-LCR (B). Corteza motora capa 4-6, 45días después de la cirugía. Escala de barras: 50μm.
 Análisis cuantitativo de la expresión de MHC-II en diversas regiones del cerebro y médula espinal, en los 3 períodos de tiempo estudiados. El grupo cELA-LCR mostró un aumento significativo del número de células microgliales, que es una evidencia clara de una etapa neuroinflamatoria. D)Imagen representativa de células positivas para MHC-II en la región lumbar. Estas células también se observaron en las proximidades de las neuronas motoras. Escala: 50 m; * p<0,05; los datos se muestran como media ± DE.")
A-C) Análisis cuantitativo de la expresión de MHC-II en diversas regiones del cerebro y médula espinal, en los 3 períodos de tiempo estudiados. El grupo cELA-LCR mostró un aumento significativo del número de células microgliales, que es una evidencia clara de una etapa neuroinflamatoria. D)Imagen representativa de células positivas para MHC-II en la región lumbar. Estas células también se observaron en las proximidades de las neuronas motoras. Escala: 50 m; * p<0,05; los datos se muestran como media ± DE.
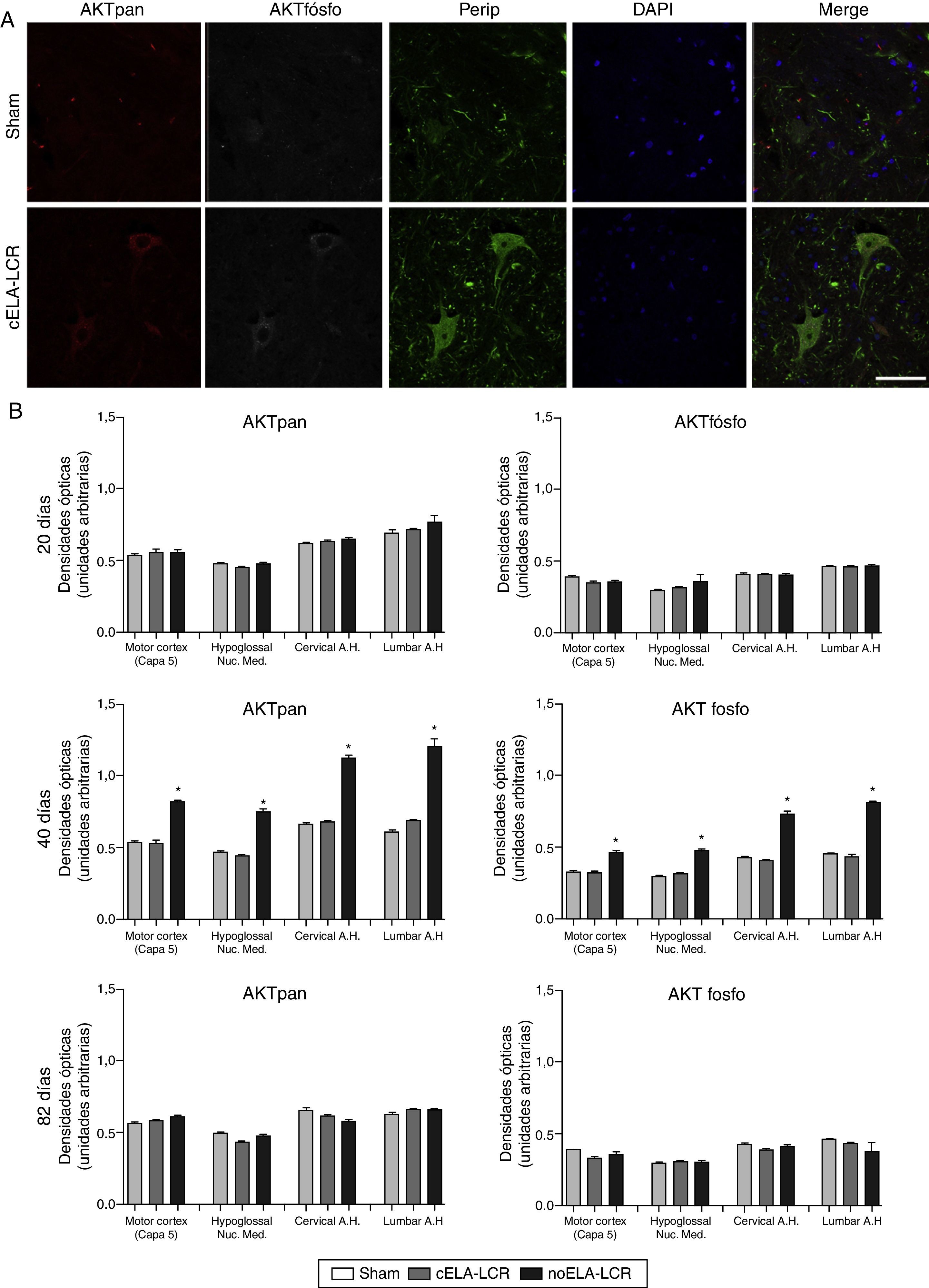
Con el fin de comprender si el incremento de caspasa-3 se acompañaba de un aumento en la expresión de proteínas estrechamente relacionadas con la supervivencia y la resistencia a la apoptosis, tales como fosfoAKT y panAKT, se llevó a cabo su cuantificación. Básicamente, el grupo de tratamiento simulado y el grupo no-ELA-LCR no mostraron cambios significativos a lo largo del estudio (20, 45 y 82días). Sin embargo, en el grupo de cELA-LCR se encontraron incrementos significativos de panAKT y fosfoAKT a los 45días, con una expresión inmunohistoquímica que era 3 veces mayor que los niveles observados en los otros grupos, pero a los 82días después de la cirugía estos datos eran similares al grupo de tratamiento simulado (fig. 4).
 y fosfoAKT (AKTfosfo) en varias regiones del sistema nervioso central de los 3 grupos de ratas. A)Imágenes de microscopia confocal de la expresión de panAKT, fosfoAKT y periferina (perip). B)Análisis de fosfoAKT y panAKT de cuantificación de densidades ópticas. Se observaron mayores expresiones de AKTpan y fosfoAKT a los 45días en el grupo de cELA-LCR (A y B). Un fenómeno interesante en el grupo de cELA-LCR fue el incremento en la expresión de periferina, fosfoAKT y panAKT en las neuronas (A). En el día 82 después de la cirugía, las concentraciones de AKT (pan y fosfo) fueron similares a los de los grupos sham y no ELA-LCR, la expresión se había regresado a niveles similares a los observados en los otros grupos a los 82días de la cirugía días post (B). Escala: 50μm.")
Expresión de panAKT (AKTpan) y fosfoAKT (AKTfosfo) en varias regiones del sistema nervioso central de los 3 grupos de ratas. A)Imágenes de microscopia confocal de la expresión de panAKT, fosfoAKT y periferina (perip). B)Análisis de fosfoAKT y panAKT de cuantificación de densidades ópticas. Se observaron mayores expresiones de AKTpan y fosfoAKT a los 45días en el grupo de cELA-LCR (A y B). Un fenómeno interesante en el grupo de cELA-LCR fue el incremento en la expresión de periferina, fosfoAKT y panAKT en las neuronas (A). En el día 82 después de la cirugía, las concentraciones de AKT (pan y fosfo) fueron similares a los de los grupos sham y no ELA-LCR, la expresión se había regresado a niveles similares a los observados en los otros grupos a los 82días de la cirugía días post (B). Escala: 50μm.
La periferina juega un papel importante en el transporte axonal en las neuronas motoras espinales. Recientemente la periferina también se ha asociado con algunas neuropatologías que caracterizan la ELA esporádica, ya que se encuentra en los cuerpos Bunina37. Por lo tanto, hemos considerado relevante analizar la periferina en las neuronas motoras, ya que su expresión se relaciona con formas de ELA esporádica. Todas las secciones del grupo cELA-LCR mostraron neuronas motoras con expresión positiva de periferina (figs. 5 y 6), pero en los animales sacrificados a los 45días se observaron niveles 4 veces más altos que en los de los otros grupos y los puntos de tiempo analizados (fig. 6). Por otra parte, las células con más inmunorreactividad de periferina también co-expresaron con panAKT y fosfoAKT. Estas células mostraban el marcador de periferina con un patrón filamentoso, que se observa también en las condiciones normales; sin embargo, en el grupo de cELA-LCR también se observaron células que contenían el marcador de periferina en inclusiones citoplasmáticas que recuerdan estructuras rugosas o granulares, que posiblemente podrían desencadenar la formación de precipitados o inclusiones de proteínas (fig. 5).
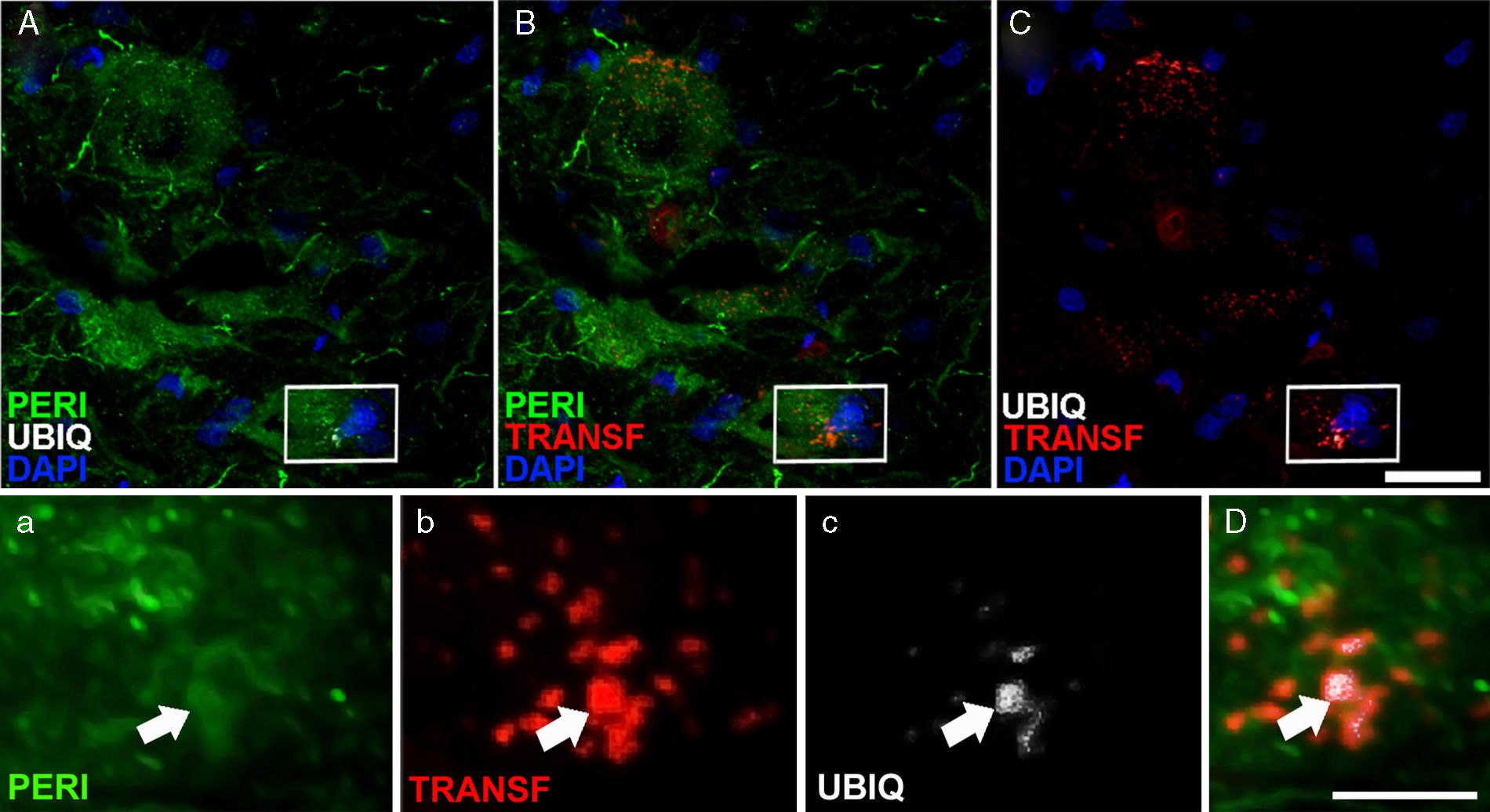
, ubiquitina (ubiq) y transferrina después de la infusión de cELA-LCR. A los 45 y 82días después de la cirugía los animales con tratamiento cELA-LCR mostraron inclusiones en el citoplasma y tenían marcaje positivo de ubiquitina en las neuronas. Inclusiones (flechas) de ubiquitina se colocalizaron con periferina y, ocasionalmente, con transferrina. Escala A-C: 20μm; a-c y D: 2μm.")
Expresión de periferina (peri), ubiquitina (ubiq) y transferrina después de la infusión de cELA-LCR. A los 45 y 82días después de la cirugía los animales con tratamiento cELA-LCR mostraron inclusiones en el citoplasma y tenían marcaje positivo de ubiquitina en las neuronas. Inclusiones (flechas) de ubiquitina se colocalizaron con periferina y, ocasionalmente, con transferrina. Escala A-C: 20μm; a-c y D: 2μm.
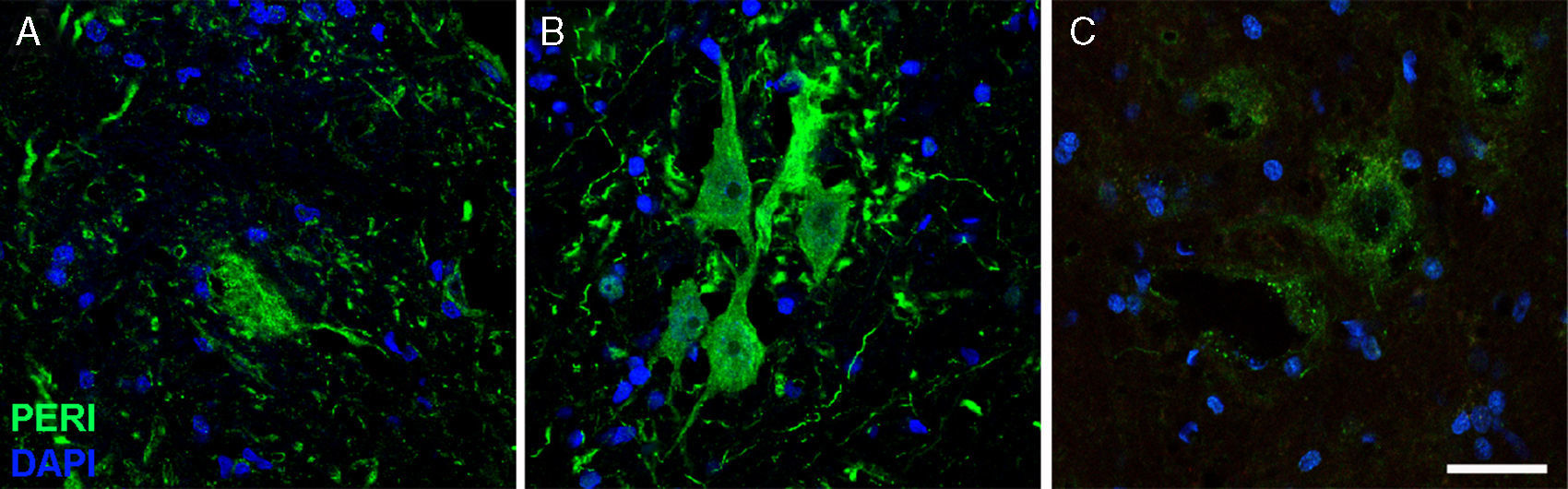
 a lo largo del curso del estudio en el grupo de cELA-LCR (A: 20días; B: 45días; C: 82días). Se observó un aumento temporal de periferina en las neuronas motoras en el día 45, que había regresado a condiciones basales al día 82. El aumento de expresión de periferina se ha asociado con acontecimientos de lesión en columna, mientras que la expresión es baja en condiciones normales. Escala: 40μm.")
Imágenes de inmunofluorescencia que muestran la expresión de periferina (peri) a lo largo del curso del estudio en el grupo de cELA-LCR (A: 20días; B: 45días; C: 82días). Se observó un aumento temporal de periferina en las neuronas motoras en el día 45, que había regresado a condiciones basales al día 82. El aumento de expresión de periferina se ha asociado con acontecimientos de lesión en columna, mientras que la expresión es baja en condiciones normales. Escala: 40μm.
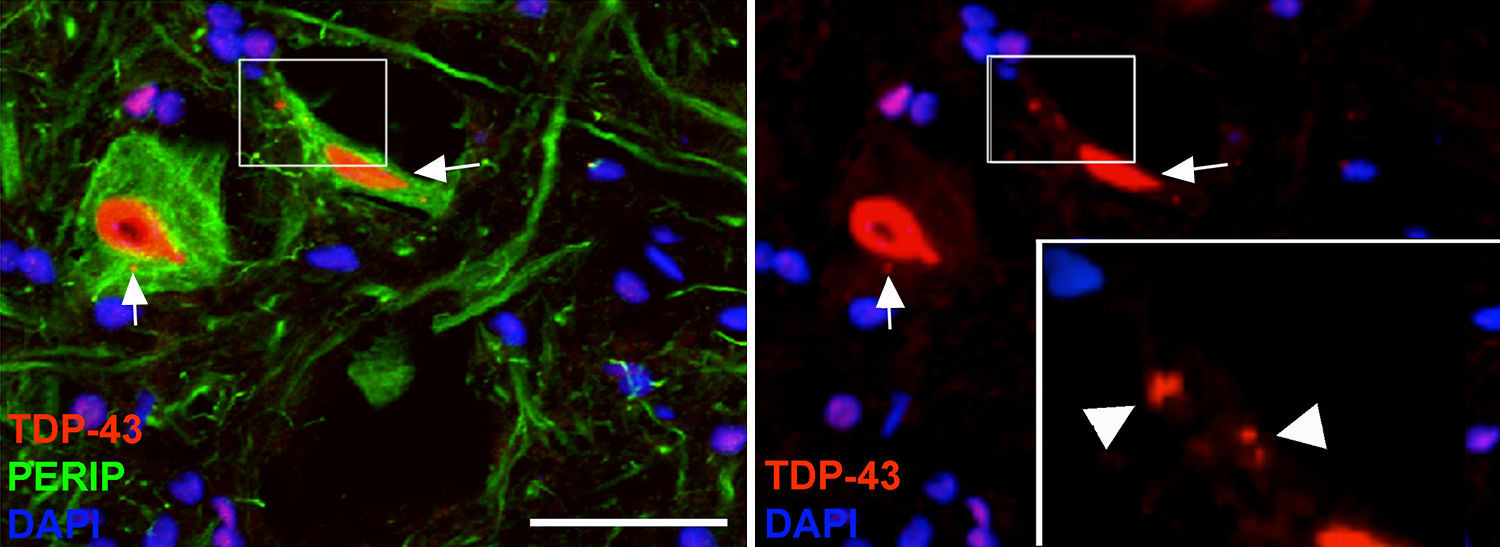
En el grupo de cELA-LCR, a los 45 y 82días después de la cirugía se observaron células con una mayor expresión de la ubiquitina, transferrina (fig. 5) y cistatinaC. Estos datos están estrechamente vinculados a formas esporádicas de ELA38. Por otra parte, se observó que había una translocación de TDP-43 en el citoplasma de las neuronas motoras (fig. 7). Curiosamente, el análisis inmunohistoquímico por microscopia confocal reveló inclusiones de TDP-43, ubiquitina y cistatinaC, y estas proteínas se colocalizaron en el citoplasma de las neuronas motoras. El incremento de estas inclusiones a los 82días fue acompañado por una disminución en la expresión de periferina (fig. 6).
Metalotioneínas. En el detalle se observan las inclusiones (cabezas de flecha). Escala: 40μm.")
Como algunos estudios han sugerido que la homeostasis del cobre y del cinc podría contribuir a la patogénesis de la ELA, se estudió la expresión de metalotioneína39,40. La expresión de metalotioneínas estaba relacionada con el curso temporal de los cambios después de la cirugía (45 y 82días) y solo se produjo en el grupo de cELA-LCR, en el que se observó en las neuronas motoras que co-expresaban ubiquitina y transferrina. El aumento de la S100B medido a los 45 y 85días después de la cirugía estaba estrechamente relacionado con el aumento de la periferina en las proximidades de las neuronas motoras. Las células que expresan S100B correspondieron al fenotipo de astrocitos (GFAP+) de neutrófilos adyacentes a neurona motora. Los astrocitos que expresan S100B están estrechamente relacionados con el equilibrio de calcio, cinc y cobre. S100B es una proteína de unión a calcio expresada exclusivamente por las células gliales.
DiscusiónEl presente estudio tiene como objetivo determinar las consecuencias de la administración sostenida ELA-LCR en ratas a fin de comprender los efectos que podrían ocurrir en pacientes que presentan citotoxicidad en su LCR. Esto demuestra que la infusión ICV sostenida de cELA-LCR en ratas produce cambios citohistoquímicos en cerebro y médula espinal que se asemeja a los encontrados en pacientes con ELA esporádica. Esos cambios no se observaron en ratas infundidas con solución salina o en animales infundidos con no-ELA-LCR. La infusión se realizó durante 20 o 43días y los tejidos se analizaron a los 20, 45 y 82días después del implante de la cánula. Los animales del estudio que se sacrificaron a los 82días recibieron la infusión durante los primeros 43días solamente.
Los cambios relevantes en las células a los 45días fueron una activación microglial seguida por astrogliosis. La activación y la proliferación de microglía antes de la aparición de los signos clínicos también se producen en pacientes con ELA41, así como en ratones transgénicos SOD1-G93A42. Además, en los ratones transgénicos ELA la microglía activada aparece antes de la aparición de la pérdida neuronal43. Esto se mantiene en línea con la observación de que la expresión reducida de SOD1-G93A en las células microgliales ralentiza la progresión de la enfermedad y prolonga la supervivencia44. Por el contrario, las células microgliales que expresan SOD1-G93A presentan aumento de neurotoxicidad y aumento en la producción de citocinas, NO y MCP-1 tras su activación in vitro45. Estos hallazgos apoyan la idea de que la activación de microglía puede jugar un papel relevante en el desarrollo de ELA46,47. A los 45días después del implante (43días de infusión con ELA-LCR) encontramos células de microglía en contacto cercano con las neuronas de la médula espinal. Estas células mostraban la morfología típica de la activación concomitante con la expresión aumentada de MHC-II, que se considera que es un marcador de la respuesta inflamatoria dependiente del complemento48,49. La astrogliosis está claramente presente, a pesar de que aún se desconoce su implicación en la patogénesis de la ELA y si aparece antes o después de la activación microglial. Otro hallazgo notable fue la sobreexpresión de S100B en astrocitos a los 45 y 82días. Esta proteína se ha correlacionado con la supervivencia en ELA50,51 y es regulada en astrocitos y neuronas motoras de la médula espinal en los pacientes52, así como en ratas infundidas durante 48h con ELA-LCR53. Los astrocitos con inclusiones están presentes también en los ratones mutantes SOD154. Asimismo, se observó que la astrogliosis y la sobreexpresión de GFAP aparecieron después de la activación de microglía. Esto podría apoyar la idea de un mecanismo de neuroinflamación como un evento temprano en la patogénesis de la enfermedad.
En los últimos años se ha puesto énfasis en la translocación citoplasmática de TDP-43, una proteína codificada por el gen cuyas mutaciones de TARDBP se han asociado con ELA4. Hemos observado que TDP-43 se encuentra en inclusiones de ubiquitina en la ELA esporádica, pero no en los casos SOD1 mutado. La presencia citosólica de TDP-43, colocalizada con ubiquitina, a los días 45 y 82días en el grupo de cELA-LCR apoya la idea de que estos cambios están relacionados con la ELA esporádica. Esto se ve reforzado por nuestros hallazgos de que los primeros cambios en las neuronas motoras incluyeron la sobreexpresión de la cistatinaC, transferrina y periferina, 3 proteínas que están estrechamente vinculadas a los cuerpos Bunina en la ELA esporádica37,55,56. De hecho, las mutaciones de periferina ya se han asociado con la ELA esporádica57.
Otra variable que se valoró fue la vía de señalización PI3K/AKT relacionada con la supervivencia celular58. Hemos hallado un aumento en la expresión de panAKT y fosfoAKT al día 45 en ratas tratadas con cELA-LCR. Este fue un hallazgo interesante teniendo en cuenta que periferina es también un sustrato de PI3K. Vale la pena señalar que el aumento de la expresión de PI3K, pero no de AKT, se ha encontrado en la médula espinal de pacientes con ELA59. Sin embargo, el incremento de fosfoAKT se ha observado en las médulas espinales de ratones SOD1-G93A en la fase asintomática60. Por otra parte, los estudios in vitro indican que la PI3K/AKT se altera en células procedentes de ratas mutadas SOD1-G93A61. Por lo tanto, la periferina y la vía PI3K/AKT podrían aumentar como una respuesta al estrés celular causado por cELA-LCR. El aumento de caspasa-3 como un signo de la apoptosis en estas ratas es compatible con este punto de vista y con el hallazgo de las neuronas motoras apoptóticas en la ELA62-64.
El último hallazgo que debe mencionarse es el incremento en la expresión de la metalotioneína en ratas infundidas con cELA-LCR. Esto está de acuerdo con informes de niveles más altos de las metalotioneínas en ratones mutados SOD165-67; un aumento similar en su ARNm se ha descrito también en estos ratones68. En consistencia con esto es la observación de que en ratones que proceden del cruce de roedores con ELA y deficientes de metalotioneína se acelera la progresión de la enfermedad68 y que su mayor expresión supone una mejoria69. En contraste, la expresión de metalotioneína y su ARNm está disminuida en la ELA70. Las metalotioneínas son proteínas Cu/Zn-vinculantes que reducen los productos reactivos del oxígeno y contrarrestan la toxicidad por metales. En este contexto, la búsqueda de una mayor expresión metalotioneína podría significar que una reacción neuroprotectora se desarrolla en respuesta a la infusión crónica de cELA-LCR en la rata; esto se mantiene en línea con el hallazgo, de la expresión incrementada de metalotioneína en las neuronas motoras de la médula espinal de ratones mutados con ELA, lo que trata de evitar la pérdida de neuronas e inhibe la progresión de la enfermedad71.
Los hallazgos histopatológicos descritos son compatibles con la hipótesis de que la citotoxicidad de LCR en ratas, inducida por semanas de infusión ICV prolongada de cELA-LCR, produce cambios en cerebro y médula espinal similares a los de las formas esporádicas de ELA. Nuestro estudio muestra algunas limitaciones. En primer lugar, los síntomas clínicos están ausentes en los 3 períodos estudiados (20, 45 y 82días). En los ratones SOD1-G93A-ELA, los primeros síntomas clínicos aparecen a los 90-100 días de edad72, pero los roedores con otras mutaciones pueden comenzar a mostrar síntomas clínicos más tarde38. Se han observado anomalías tempranas, como la fragmentación de Golgi a los 31días de edad73; también parece posible que se produzcan cambios sutiles antes de esa edad74. En los ratones con mutaciones SOD1, las primeras alteraciones aparecen en la médula espinal cervical38. También se han observado cambios en la médula espinal cervical y lumbar similares a los observados en la corteza motora cerebral, lo que indica que el efecto nocivo de cELA-LCR infundido en el ventrículo cerebral había propagado distalmente a la médula espinal. Esta observación es compatible con la hipótesis de que la progresión de la enfermedad podría estar mediada en parte a través del LCR. En segundo lugar, aún se detectaron varios marcadores de proteínas que se sobreexpresan a los 45días, aunque en concentraciones menores, a los 82días (es decir, periferina, GFAP, panAKT y fosfoAKT); en otros se sobreexpresa por igual a los 45 y 82días (es decir, la cistatinaC, transferrina, ubiquitina y translocación TDP-43), aunque con la limitación de que los tamaños de muestra de grupos de estudio no eran similares y el número de ratas de los grupos de control fue bajo. Sin embargo, la expresión anormal de estas proteínas en el grupo cELA-LCR es clara y los diferentes resultados en los grupos de control confirman que los cambios no son debido a artefactos del método. En tercer lugar, cELA-LCR fue infundido por ICV durante 43días, que era el plazo máximo para el volumen de la minibomba. A pesar del hecho de que el cELA-LCR no fue administrado entre el día 43 y el día 82, las neuronas motoras apoptóticas solamente se observaron en animales examinados en el día82. Esto sugiere que el proceso nocivo podría permanecer activo después del día 43, y por tanto parece posible que la extensión de la infusión de cELA-LCR más allá de 43días y/o al siguiente curso temporal de la enfermedad más allá de 85días podría revelar las manifestaciones clínicas de una neurona motora parecida a las de las formas esporádicas de ELA.
En conclusión, este estudio presenta las primeras consecuencias de citotoxicidad LCR-ELA después de semanas de infusión ICV sostenida con LCR de pacientes con ELA, que previamente se había probado que mostraban efectos citotóxicos en un cultivo primario de neuronas motoras. A los 45 y 82días después del inicio de la infusión la corteza motora, el tálamo y la médula espinal cervical-lumbar mostraron varios cambios en los marcadores típicos de la enfermedad de ELA humana. Estos hallazgos pueden mejorar nuestra comprensión de la patogénesis y la evolución de la ELA y ayudar a encontrar nuevas dianas terapéuticas útiles para el desarrollo de nuevos compuestos que retrasen o incluso puedan llegar a detener la progresión de la enfermedad.
FinanciaciónEste estudio fue apoyado por cuatro subvenciones concedidas a JMG por la Mutua Madrileña, España, en 2008 y 2009. También fue apoyado por las siguientes subvenciones concedidas a AGG por instituciones españolas: 1) NDE07/09, Agencia Laín Entralgo, Comunidad de Madrid; 2) Fundación Eugenio Rodríguez Pascual; 3) Fundación CIEN, Instituto de Salud CarlosIII; 4) RENEVAS-RETICS-RD06-026, Instituto CarlosIII; 5) SAF 2010 a 21795, Ministerio de Economía y Competitividad, España, y 6) MAT2011-28791-C03.
Conflicto de interesesLos autores declaran que no existen conflictos de intereses.
AgradecimientosLos autores desean agradecer la colaboración de Pablo González y M. Cruz Rodríguez por la coordinación de los procedimientos de cuidado de los animales, y a la Fundación Teófilo Hernando por su continuo apoyo.

