



Nemaline myopathy (NM) is a congenital myopathy that follows an autosomal dominant or recessive inheritance pattern. It is classified according to the age of onset and the severity of respiratory and muscle involvement. From an anatomical pathology viewpoint, NM is characterised by deposition of rod-like cytoplasmic inclusions (nemaline rods) that stain red on Gomori trichrome. These rod-like structures are mainly composed of α-actin and have been located at the level of the Z-band of the sarcomere in electron microscopy studies.1,2
In adults, NM may either be hereditary, with a slowly progressive course and symptomaticity in adulthood, or present in adulthood and progress subacutely, without a hereditary pattern. This sporadic adult-onset form, known as sporadic late-onset NM (SLONM), is characterised by onset after the age of 40 years and is frequently associated with monoclonal gammopathy of undetermined significance (MGUS) or may be diagnosed in the context of HIV infection.3–7 A potentially fatal disease, SLONM constitutes a challenge for neurologists as this myopathy may be treatable, according to the available evidence on the response to immunotherapy.
We present the case of a patient with respiratory failure who presented nemaline rods in a muscle biopsy.
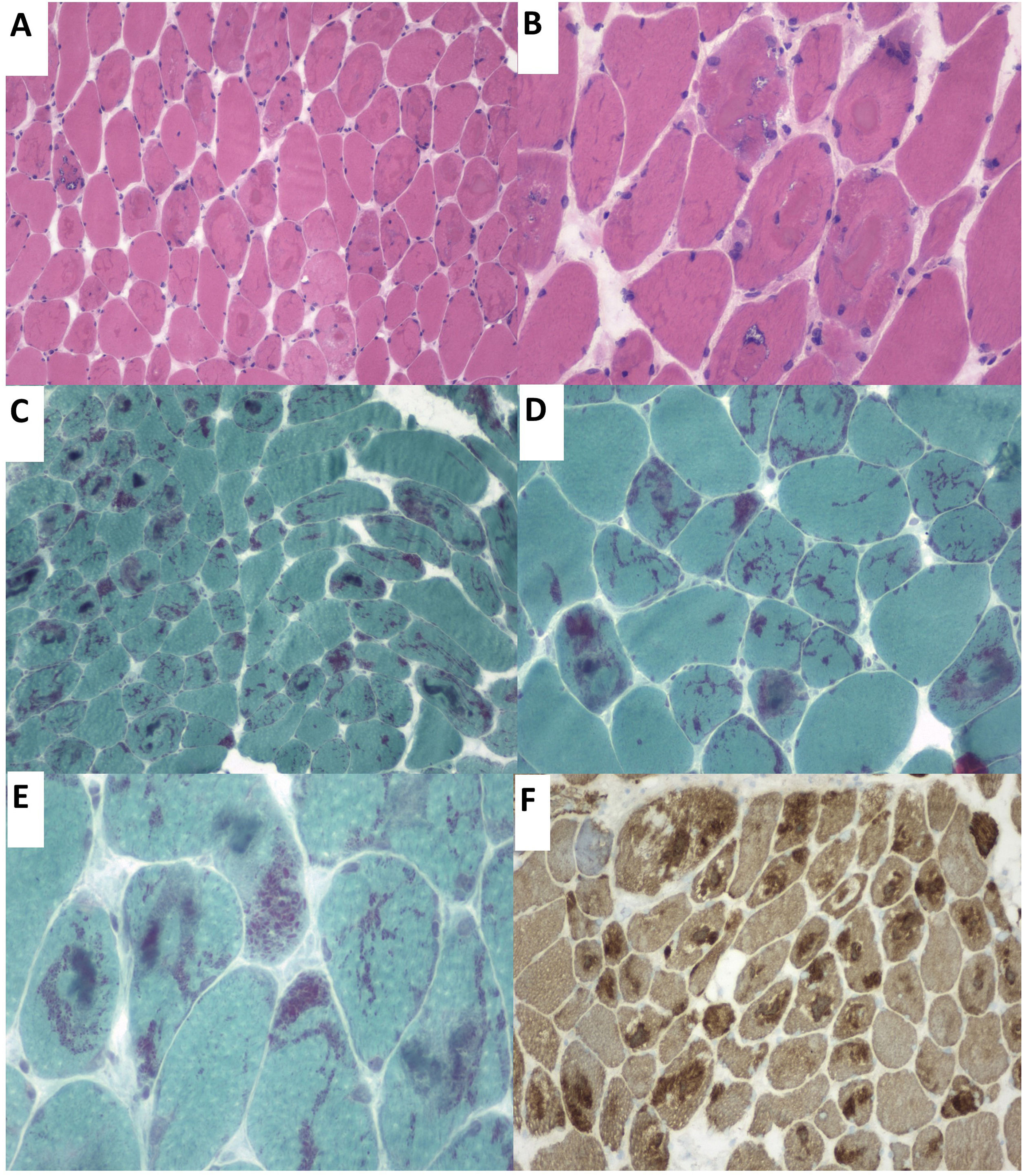
Our patient was a 57-year-old woman who was admitted to the pulmonology department due to hypercapnic respiratory failure requiring non-invasive mechanical ventilation (NIMV). She had presented sensorineural hearing loss since childhood. Questioned specifically about muscle function, she reported a 6-month history of proximal muscle weakness in the lower limbs. The neurological examination detected bilateral ptosis, neck extensor muscle weakness, and proximal limb muscle weakness (muscle strength 4/5). A laboratory analysis revealed a normal-to-low creatine kinase level (22–44U/L). The neurophysiological study revealed a myopathic pattern in proximal muscles, with low-amplitude, polyphasic motor unit potentials and isolated positive sharp waves. Neurography and repetitive nerve stimulation detected no alterations. A biopsy of the deltoid muscle using the Gomori trichrome stain identified nemaline rods in most muscle fibres (Fig. 1).
 Haematoxylin-eosin stain. Presence of rimmed vacuoles (left) and numerous muscle fibres with irregular eosinophilic sarcoplasmic inclusions occupying a large part of the fibres. (B) Close-up of the image displayed in A. (C and D) Gomori trichrome stain displaying red-staining nemaline structures. (E) Close-up image of rod-like nemaline structures. (F) The immunohistochemical study showed nemaline rods containing actin filaments.")
(A) Haematoxylin-eosin stain. Presence of rimmed vacuoles (left) and numerous muscle fibres with irregular eosinophilic sarcoplasmic inclusions occupying a large part of the fibres. (B) Close-up of the image displayed in A. (C and D) Gomori trichrome stain displaying red-staining nemaline structures. (E) Close-up image of rod-like nemaline structures. (F) The immunohistochemical study showed nemaline rods containing actin filaments.
Serum protein electrophoresis and immunoelectrophoresis did not detect monoclonal protein, and a serology test for HIV infection yielded negative results. A genetic panel for congenital myopathies ruled out mutations in the genes known to cause NM. The study incidentally detected a mutation, c.1229G>A; p.(Arg410His), in heterozygosis in the MYH14 gene, located on chromosome 19. The patient was initially treated with intravenous immunoglobulins and subsequently started maintenance treatment with prednisone. She remains clinically stable at 18 months of follow-up but continues to require NIMV overnight.
SLONM is a rare muscle disorder with a heterogeneous clinical presentation; clinical onset as respiratory failure in adults is infrequent.8,9 In nearly half of cases, SLONM is associated with presence of haematological disease, mainly MGUS and multiple myeloma, as well as HIV infection. HIV-related NM is characterised by absence of facial or respiratory involvement and favourable clinical response to immunosuppressive treatment.4 SLONM associated with monoclonal protein has traditionally been considered to present poorer prognosis, and is characterised by severe weakness and muscle atrophy, dysphagia, and respiratory failure. However, recent evidence suggests that these patients may respond to intensive treatment with intravenous immunoglobulins followed by chemotherapy or autologous stem cell transplantation.6,7,10
Patients with SLONM do not present known mutations in NM-related genes. MYH14 encodes non-muscle myosin heavy-chain, which is expressed across all tissues but mainly in skeletal muscle (the term “non-muscle myosin” aims to differentiate this ubiquitous form of myosin from muscle tissue–specific myosin). MYH14 is associated with hereditary nonsyndromic sensorineural hearing loss. The MYH14 mutation detected in our patient caused hearing loss, although MYH14 mutations have been linked to complex phenotypes combining hearing loss with myopathy and peripheral neuropathy, following an autosomal dominant inheritance pattern.11 The patient did not consent to familial cosegregation analysis; her mother also presented sensorineural hearing loss. Currently, this genetic variant must be considered to be of uncertain significance for NM. However, as further analysis may establish an association between this mutation and our patient’s phenotype and anatomical pathology findings, we cannot be certain that the case presented here results from the incidental copresence of 2 rare entities.
The pathogenesis of SLONM is currently believed to have an inflammatory/autoimmune basis,12 and the few case series published to date suggest that immunosuppressive treatment may be suitable in all patients.6,10,12
