



SYNGAP1 mutation was first described in 2009 in patients with nonsyndromic psychomotor retardation and autistic spectrum disorders. Subsequently, in 2013, it was reported to cause developmental and epileptic encephalopathy (DEE).1 The gene is located on chromosome 6p21.32 and encodes the synaptic Ras GTPase-activating protein 1 (SYNGAP1), regulating the excitation/inhibition balance of hippocampal neurons in association with N-methyl D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. The majority of mutations are de novo mutations, causing a truncated, hypofunctional protein.1,2 Up to 98% of patients present epilepsy,1,3 showing drug resistance in up to 50% of cases in some series,4 with the most frequently described seizures being atypical absence seizures, eyelid myoclonus, myoclonic-atonic seizures with falls, and reflex seizures mainly triggered by eating.1
Cannabidiol is an approved drug in Europe for treating Lennox–Gastaut syndrome and for Dravet syndrome as an adjuvant treatment with clobazam, in patients older than 2 years, and is approved in the USA for treating tuberous sclerosis complex.5 It acts by reducing neuronal hyperexcitability, and also presents anxiolytic and sleep-regulating effects.6 Some studies have shown cognitive and behavioural improvements in animal models and human trials,7 and effectiveness in other conditions associated with epileptic seizures.8
We present the case of a patient diagnosed with encephalopathy associated with SYNGAP1 mutation at our centre, who was treated with cannabidiol due to drug resistance, achieving a significant clinical improvement.
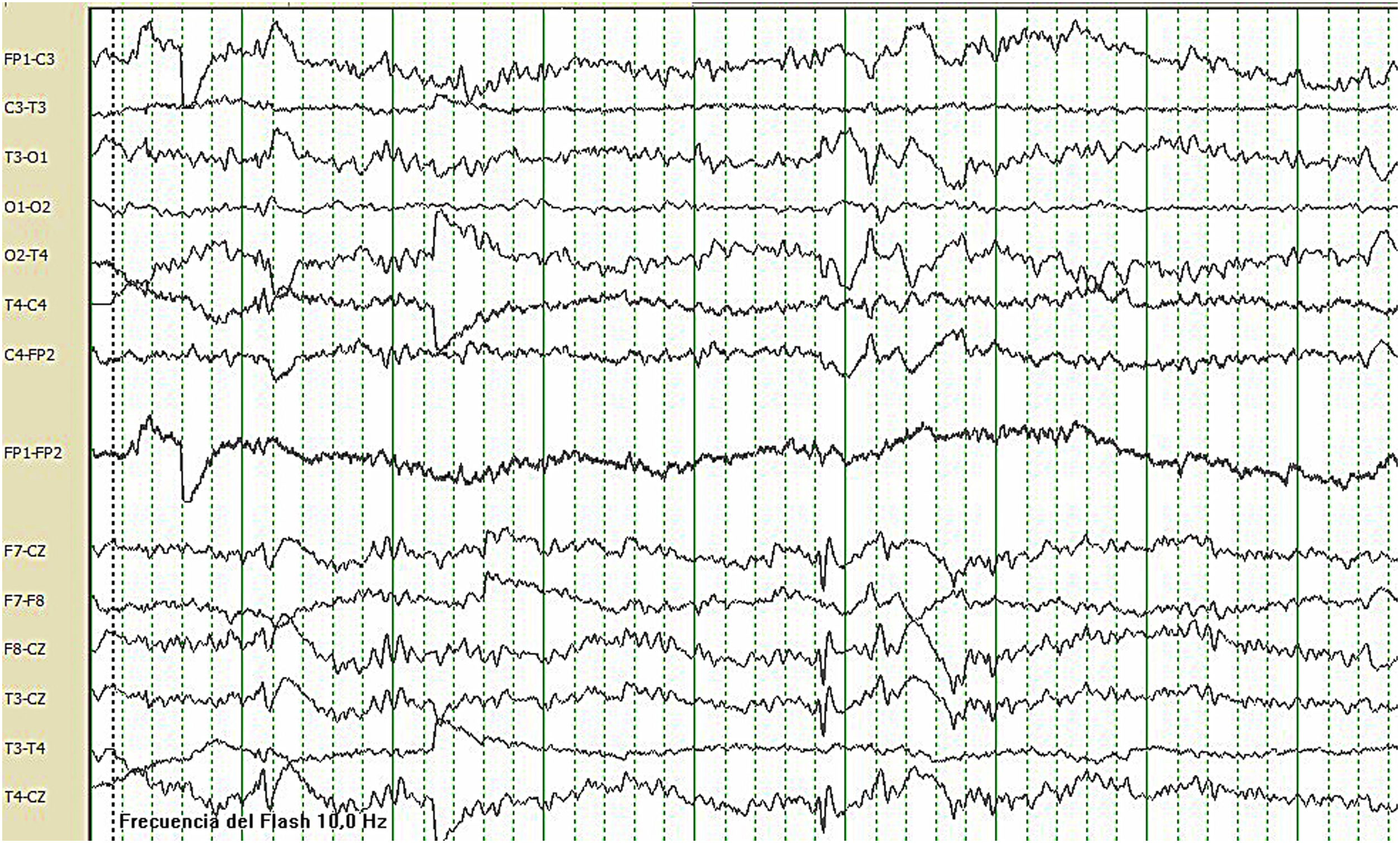
Case reportWe present the case of a 21-year-old woman with no relevant perinatal medical history. From the first days of life, she presented daily episodes of disconnection from the environment, with oral automatism (swallowing), episodes compatible with palpebral and axial myoclonia, and atonic seizures, accompanied by psychomotor retardation. Subsequently, she developed generalised tonic–clonic seizures (GTCS) and gelastic seizures. The patient never acquired language and was diagnosed with autism spectrum disorder, developing episodes of behavioural alterations. Karyotyping, a brain MRI study, and genetic and metabolic studies all yielded normal results. The patient started attending consultations at the adult epilepsy unit at Hospital Regional Universitario de Málaga (HRUM), and showed focal seizures with secondary generalisation and atonic seizures several times per day, in addition to GTCS twice weekly and frequent myoclonus. Several combinations of antiepileptic drugs (AED) were tried, including carbamazepine, perampanel, brivaracetam, and clonazepam, which were suspended due to ineffectiveness; therefore, the patient met criteria for drug resistant epilepsy. She was receiving treatment with valproic acid (900mg daily) and lacosamide (400mg daily). A video-EEG monitoring study recorded no ictal events but did detect background slowing activity compatible with diffuse encephalopathy with high-voltage rhythmic delta waves in bilateral central regions and vertex (Fig. 1). In 2018, an exome sequencing study was performed, revealing heterozygous presence of a de novo nonsense mutation of the SYNGAP1 gene (c.1861C>T), resulting in replacement of the amino acid Arg621, truncating the protein.

Given the lack of seizure and behavioural control, we decided to start treatment with cannabidiol (300mg every 12h) and clobazam (10mg at night). At 3 months, we observed the effectiveness of the new treatment, with seizure frequency decreasing by more than 50% and a reduction in seizure duration, with atonic seizures disappearing. The effectiveness of the rescue medication also seemed to have improved. Furthermore, improved seizure control led to an improvement in functional status, with the patient recovering the ability to walk autonomously and showing better behavioural control.
DiscussionDEE associated with SYNGAP1 mutation is a recently described, rare disease, which presents with a characteristic epileptic syndrome characterised by absence seizures, palpebral myoclonus, and myoclonic-atonic seizures. We present the case of a patient with DEE secondary to a pathogenic mutation of the SYNGAP1 gene, which was described in 2020,9 and who, as a peculiar feature, presented gelastic seizures. We are aware of no other case presenting this type of seizures. Another surprising aspect of our patient is her good response to cannabidiol treatment. Mutations in this gene induce an increase in levels of the protein transient receptor potential cation channel subfamily V member 1 (TRPV1), one of the mechanisms causing the excitation/inhibition imbalance, and cannabidiol seems to induce a rapid activation and desensitisation of this receptor.8 In fact, some published series report a good clinical response.1,10 Based on these data, we decided to prescribe compassionate use of cannabidiol, achieving a clinical improvement, both in terms of seizure control and at the cognitive and behavioural levels.
As cases are increasingly being reported in the literature and genetic studies are becoming more widely available, in the coming years we will probably be able to identify a greater number of patients, which will enable us to better characterise the disease and obtain data regarding treatment effectiveness. Compassionate use of cannabidiol may be an effective treatment for DEE, but further studies are needed on this matter.
