



Lissencephaly is a brain malformation affecting the cerebral cortex, and is caused by a defect in neuronal development and migration.1,2 In patients with lissencephaly, neurons are unable to migrate to the surface layers of the cortex, resulting in fewer convolutions, and accumulate in deep layers of the subcortical white matter, forming a subcortical layer of grey matter (band heterotopia). Lissencephaly has classically been divided into 2 types: type 1 is characterised by complete absence of gyri (agyria), which results in a smooth brain surface, or incomplete gyral formation, leading to broader gyri (pachygyria); type 2 features disorganised collections of neurons and loss of normal cortical lamination. Type 2 is associated with several genetic syndromes, including Fukuyama type congenital muscular dystrophy, Santavuori congenital muscular dystrophy, and Meckel-Gruber syndrome.3 Lissencephaly may be associated with malformations of the central nervous system or other systems, for example in lissencephaly with cerebellar hypoplasia (LCH),1,2,4–6 or with certain genetic syndromes, including Walker-Warburg syndrome.3 LCH is reported to be linked to mutations in the RELN and TUBA1A genes. RELN, located on the long arm of chromosome 7 (7q22), encodes reelin, a protein involved in neuronal migration, synaptic plasticity, and transmission of nerve impulses. At least 6 autosomal recessive mutations in RELN have been described. TUBA1A, located on the long arm of chromosome 12 (12q13.12), encodes alpha-tubulin; this protein is a structural constituent of microtubules, which play a major role in cell division and movement. Ten autosomal dominant TUBA1A mutations have been described; these have been found in 30% of patients with LCH.1
Extrahepatic biliary atresia (EBA) is classified into 3 types depending on whether the condition presents in isolation (type 1), in association with other congenital defects but without being considered a polymalformative syndrome (type 2), or as a polymalformative syndrome (type 3).7,8 Patients with type 2 and 3 EBA present cardiac, gastrointestinal, spleen, and genitourinary abnormalities.
The case presented here was particularly challenging due to the presence of neonatal seizures secondary to a malformation of cortical development and fatal liver failure. This is the first reported case of an association between LCH and EBA.
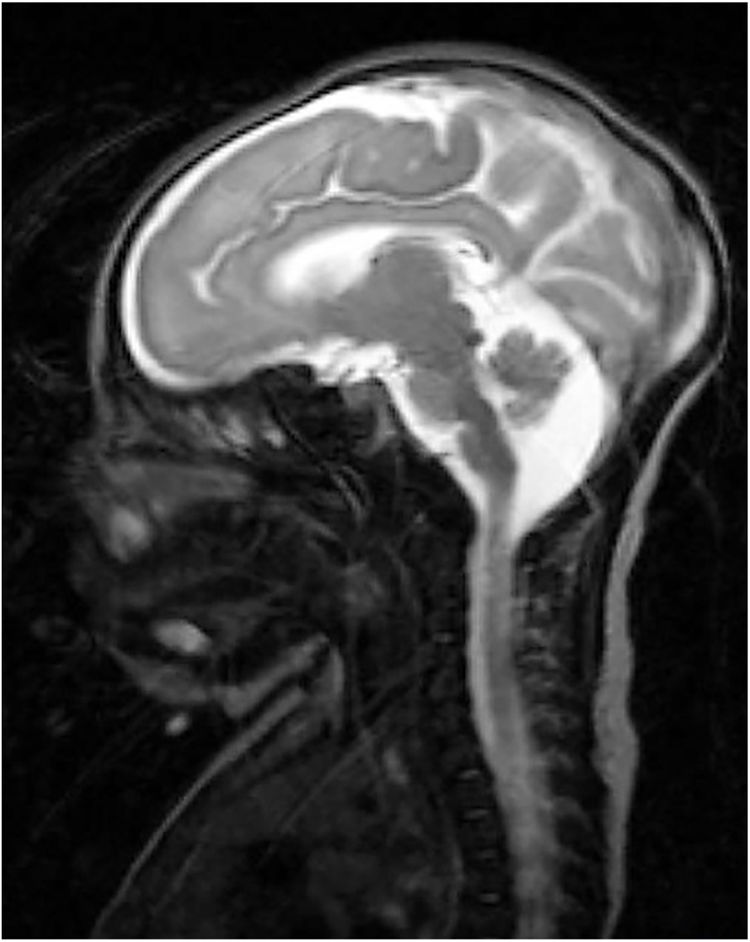
Our patient was a full-term neonate with intrauterine growth retardation from week 34. The pregnancy was uneventful. The parents were nonconsanguineous and the baby was born by normal vaginal delivery. Birth weight was 2470 g (P2; –2.18 SD), length was 43 cm (P2; –2.3 SD), and head circumference was 30 cm (< P1; –3.21 SD). Two hours after birth, the child started to make sucking motions, which were subsequently associated with clonic movements of the face and left arm, lasting 4 min. The patient presented similar episodes during the first 24 h of life, which resolved with phenobarbital. We performed an aetiological study of neonatal seizures. A blood count revealed thrombocytosis and slightly elevated thyroid hormone levels. An electroencephalography study detected no alterations. Transfontanellar ultrasound revealed thinning of the corpus callosum and mega cisterna magna. In addition to these findings, a brain MRI scan (Fig. 1) also revealed partial atrophy of the cerebellar vermis and moderate sulcal underdevelopment in the frontal and occipital regions, compatible with LCH. The patient received treatment with phenobarbital, levetiracetam, and clobazam due to poor seizure control. A month and a half after birth, a follow-up blood test revealed cholestasis (total bilirubin 6.8 mg/dL, conjugated bilirubin 5.3 mg/dL, gamma-glutamyl transferase 2120 IU/L). An analysis of alpha-1 antitrypsin levels, a study of hormonal and metabolic function and infections, and an abdominal ultrasound revealed no abnormalities. We suspected EBA and decided to administer conservative treatment due to the patient’s poor prognosis. A genetic study of lissencephaly and congenital disorders of glycosylation yielded negative results. At 7 months of life, the patient was brought to hospital due to fever and decompensation of the underlying liver disease; despite medical treatment, she died due to cardiorespiratory arrest. A post-mortem liver biopsy confirmed the diagnosis of EBA. This is the first case of LCH associated with EBA to be reported in the literature. To date, no study has described mutations linking LCH and EBA.
Funding
This study has received no funding of any kind.
Please cite this article as: del Castillo Velilla I, Martínez Jiménez MD, Martín MP, García Cabezas MA. Lisencefalia, hipoplasia cerebelar y atresia de vías biliares extrahepáticas: una asociación inusual. Neurología. 2020;35:502–503.
