



La glucosilación de las proteínas es fundamental en el correcto funcionamiento de numerosos procesos biológicos, como el plegamiento y la estabilidad de las proteínas, la unión a receptores intracelulares, la comunicación intracelular, etc.1. Las mutaciones que afectan la codificación de alguna de las proteínas involucradas en este proceso provocan los denominados defectos congénitos de la glucosilación de las proteínas (conocidos por sus iniciales en inglés como CDG, de congenital disorders of glycosylation), que están clasificados en diferentes subtipos, dependiendo del punto en el que se altere el proceso. La mayoría de los CDG son enfermedades multisistémicas que afectan a varios órganos y sistemas. Lo más habitual es que los pacientes DPAGT1-CDG (MIM 608093) presenten retraso psicomotor, enfermedad neuromuscular, anomalías endocrinológicas y rasgos dismórficos2.
Por otro lado, los síndromes miasténicos congénitos (SMC) son enfermedades provocadas por mutaciones en genes que codifican proteínas que son esenciales para el mantenimiento de la integridad de la transmisión neuromuscular3–5. Estos pacientes presentan debilidad que empeora con la fatiga, pero la edad de inicio, la distribución de la debilidad y la respuesta al tratamiento son variables3–5. Mutaciones en ALG2, ALG14, GFPT1, GMPPB5 y DPAGT1 (MIM 614750) pueden provocar casos de SMC con debilidad de predominio proximal, con escasa afectación facial y ocular6. Las mutaciones en DPAGT1 se habían clasificado tradicionalmente como un defecto congénito de glucosilación7–9, que cursaban con fallo de medro, hipotonía grave, microcefalia, crisis epilépticas refractarias y discapacidad intelectual. Todos los casos fallecieron en el primer año de vida9. La grave hipotonía que presentaban estos pacientes evidencia que la función neuromuscular estaba afectada. Sin embargo, otro grupo de pacientes presentan síntomas de miastenia congénita sin el resto de los síntomas de DPAGT1-CDG9.
Exponemos el caso de un paciente de 10 años con 2 mutaciones(heterocigoto compuesto) en DPAGT1, que presenta un cuadro de encefalopatía con rasgos de trastorno del espectro autista y síndrome miasténico congénito (con afectación muscular proximal, sin sintomatología ocular ni bulbar), que ha respondido satisfactoriamente a tratamiento con piridostigmina.
Varón de 10 años, remitido a los 10 meses de vida por un cuadro de síndrome dismórfico, retraso psicomotor e hipotonía. No se referían antecedentes familiares de interés. El embarazo, el parto y el período neonatal fueron normales. El peso al nacimiento fue de 4.150g. Apgar 9/10. El cribado metabólico fue normal. En la exploración destacaban ciertos rasgos dismórficos: microcefalia relativa, frente amplia, fisuras palpebrales estrechas, mamilas separadas y distribución anómala de la grasa, de predominio en el tronco y la región proximal de las extremidades, con miembros delgados distalmente. Presentaba importante hipotonía y debilidad. Los reflejos osteotendinosos eran débiles. En cuanto al desarrollo psicomotor, fue retrasado, alcanzando el sostén cefálico a los 7-8 meses y la sedestación a los 2-3 años. No ha alcanzado la deambulación autónoma. El cuadro ha permanecido estable, sin empeoramientos, a lo largo de su evolución. Actualmente, el paciente presenta discapacidad intelectual grave, con escaso interés por el medio y ausencia de lenguaje expresivo a los 10 años. No presenta ataxia ni dismetría. No se refiere ptosis, episodios de atragantamiento ni disfagia. El patrón de debilidad afecta predominantemente a miembros superiores, presentando, cuando se pone furioso, capacidad y precisión para defenderse con los miembros inferiores.
En cuanto a las exploraciones complementarias, la bioquímica, la RM cerebral y el estudio neurometabólico fueron normales. Se le realizó un EMG a los 3 años, que mostró un patrón miopático. En la biopsia muscular se observaron signos de miopatía con desproporción de fibras musculares. No se realizó microscopia electrónica, lo cual retrasó probablemente el diagnóstico. Ante la afectación cognitiva, se le efectuó un perfil de sialotransferrinas en suero (2009), que mostraba un patrón tipo 1 compatible con defecto de la glucosilación tipo i. El diagnóstico de DPAGT1-CDG se confirmó mediante estudio genético por secuenciación masiva, identificando 2 mutaciones: c.329T>C (p.Phe110Ser) –esta no descrita previamente– y c.902G>A (p.Arg301His) en el gen DPAGT1. Posteriormente ha seguido revisiones en consultas de Cardiología, Oftalmología y Endocrinología, sin haber presentado complicaciones relevantes. La determinación de anticuerpos antirreceptor de acetilcolina y anti-MUSK fue negativa.
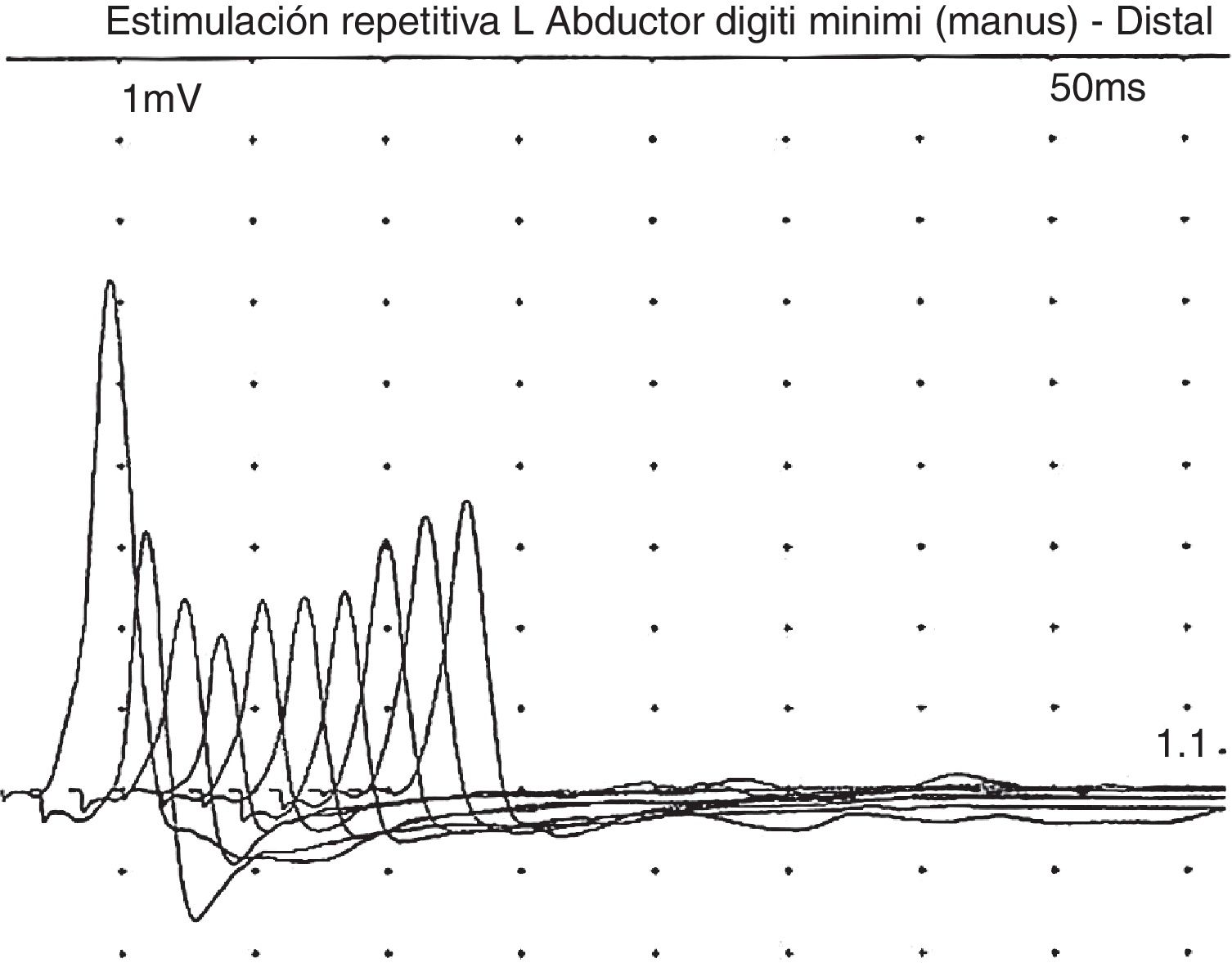
En una visita reciente, a los 10 años de vida, la familia comentó que el paciente, después de recibir fisioterapia de forma intensiva, presentaba mayor debilidad muscular (no ptosis ni sintomatología bulbar), que se recuperaba tras un período de descanso. Revisando la bibliografía, y ante la posibilidad de que asociara sintomatología miasteniforme, se le realizó EMG, pero esta vez orientando a dicha enfermedad, en el que sí se apreció claramente la respuesta decremental del 40% ante la electroestimulación repetitiva (fig. 1). Ante este hallazgo, realizamos una prueba de tratamiento con piridostigmina, tras la que el paciente presentó una importante mejoría de la fatigabilidad y de la fuerza, pudiendo ser capaz de deambular con apoyo unos metros, levantarse de la silla y mejorando su postura en sedestación. Este hecho se constató mediante la escala Myastenia Gravis Composite10, en la que se obtuvo una puntuación de 9 antes del tratamiento con piridostigmina, a expensas de debilidad moderada de cuello y proximal de hombros y caderas, y quedando tras el tratamiento con piridostigmina a 120mg/8h con una puntuación en la Myastenia Gravis Composite de 4, por debilidad leve en cadera y hombros.

En nuestro paciente, a diferencia de otros casos publicados, llama la atención la coexistencia de sintomatología muscular leve-moderada (que además fue mejorando progresivamente) y la importante afectación cognitiva, pese a lo cual el paciente ha presentado un pronóstico favorable, sin complicaciones respiratorias, epilepsia o dificultades en la alimentación. Dada la buena respuesta inicial a la piridostigmina, esperamos seguir apreciando mejoría en su calidad de vida.
La disfunción de DPAGT1 provoca un defecto de varios componentes de la unión neuromuscular: las subunidades del receptor de acetilcolina, la agrina, la cinasa específica muscular y la laminina11. Por otro lado, la cantidad del receptor de acetilcolina presente en la placa motora de los pacientes con mutaciones en DPAGT1 también estaba disminuida2. Estos cambios explican la buena respuesta a piridostigmina que suelen presentar estos pacientes. La combinación de estas alteraciones es suficiente para explicar la debilidad muscular que presentan.
En cuanto a la sintomatología, se trata de un cuadro habitualmente grave: de los 28 pacientes descritos, 23 fallecieron antes de los 5 años12. La mayoría presentaban discapacidad intelectual moderada/grave (aunque existen pacientes con inteligencia normal)13, microcefalia, hipotonía y epilepsia. Otros síntomas menos frecuentes fueron dificultad en la alimentación, apneas, insuficiencia respiratoria, anemia crónica, cataratas, hipertricosis, hipertonía de extremidades, artrogriposis, temblor, mamilas invertidas, distribución anómala de la grasa, atrofia papilar o hipoacusia neurosensorial12–14. El perfil de sialotransferrinas en suero estaba alterado en todos los casos. Entre estos 28 casos publicados, 13 de ellos presentaban síndrome miasténico congénito. En algunos pacientes se ha descrito que los síntomas pueden iniciarse más tarde, incluso a los 17 años12–14. La miastenia de estos pacientes cursa con debilidad de predominio proximal que se incrementa con el ejercicio (fatigabilidad), y suele respetar los músculos oculares y faciales12–14. La mayoría de los pacientes respondieron muy bien a piridostigmina12–14. El patrón EMG característico fue una respuesta decremental con la electroestimulación repetitiva a 3Hz11,12. La neuroimagen habitualmente fue normal.
Hasta el momento sigue sin estar claro por qué mutaciones en el mismo gen pueden dar lugar a diferentes modos de presentación. La unión neuromuscular parece ser un punto especialmente sensible a la carencia de DPAGT1, por lo que cualquier mínima reducción en su actividad puede ser suficiente para provocar sintomatología.
Respecto a nuestro paciente, habitualmente los casos de presentación tan precoz suelen tener peor evolución (fallecen en los primeros años de vida). Ante un niño con déficit cognitivo con microcefalia y debilidad muscular con escasa o nula afectación ocular o facial, cabe plantearse la posibilidad de que esta se deba a alguna mutación en DPGAT1, ya que esto puede tener implicaciones en cuanto al pronóstico y al tratamiento.

