



Alexander disease (AxD) is a type of leukodystrophy. Its pathological basis, along with myelin loss, is the appearance of Rosenthal bodies, which are cytoplasmic inclusions in astrocytes. Mutations in the gene coding for glial fibrillary acidic protein (GFAP) have been identified as a genetic basis for AxD. However, the mechanism by which these variants produce the disease is not understood.
DevelopmentThe most widespread hypothesis is that AxD develops when a gain-of-function mutation causes an increase in GFAP. However, this mechanism does not explain myelin loss, given that experimental models in which GFAP expression is normal or mutated do not exhibit myelin disorders. This review analyses other possibilities that may explain this alteration, such as epigenetic or inflammatory alterations, presence of NG2 (+) – GFAP (+) cells, or post-translational modifications in GFAP that are unrelated to increased expression.
ConclusionsThe different hypotheses analysed here may explain the myelin alteration affecting these patients; several of these mechanisms may co-occur. These theories raise the possibility of designing therapies based on these mechanisms.
La enfermedad de Alexander (AxD) es una leucodistrofia. Su base patológica, junto a la pérdida de mielina, es la aparición de los cuerpos de Rosenthal, que son inclusiones citoplasmáticas en células astrocitarias. Mutaciones en el gen que codifica la GFAP se han identificado como una base genética para AxD. Sin embargo, no se conoce el mecanismo por el cual estas variantes producen la enfermedad.
DesarrolloLa hipótesis más extendida es que AxD se desarrolla por un mecanismo por ganancia de función debido al incremento de GFAP. Sin embargo, este mecanismo no explica la pérdida mielínica, dado que los modelos experimentales que expresan GFAP normal o mutada no generan alteración mielínica. En la presente revisión se analizan otras posibilidades que permitan justificar dicha alteración, como son alteraciones epigenéticas, inflamatorias, la existencia de células NG2 (+)-GFAP (+) o cambios postraslacionales sobre la GFAP al margen de la mayor expresión.
ConclusionesLas diferentes hipótesis analizadas pueden explicar la alteración de la mielina que aparece en los pacientes y que pueden presentarse asociadas y abren la posibilidad de plantear terapéuticas basadas en estos mecanismos.
Alexander disease (AxD), named after the physician who first described it in 1949,1 is a leukodystrophy causing the destruction of myelin. In addition to myelin loss, the pathophysiology of the disease involves the formation of Rosenthal fibres,2 cytoplasmic inclusions within glial cells that have also been observed in some gliomas. These inclusions are formed by glial fibrillary acidic protein (GFAP), α–β crystallin, and heat shock protein 27 (HSP27),3,4 although other proteins such as vimentin, p62, and plectin have also been reported.5 From a clinical viewpoint, the disease has three different forms: infantile, juvenile, and adult; the infantile form has the poorest prognosis.6–8 Mutations in the GFAP gene constitute the genetic basis of AxD. These mutations, constituting changes in 32 specific nucleotides, are present in both familial and sporadic cases.9,10 However, we are yet to determine the mechanism by which these mutations cause GFAP aggregation within astrocytes, the way in which GFAP expressed by astrocytes contributes to symptoms, and especially the mechanism of demyelination. Radiology studies reveal periventricular demyelination11; patients with long survival times display extremely severe demyelination, affecting nearly all the white matter.12
GFAP, first isolated and described by Eng in 1969,13 is a component of the intermediate filaments found in astrocytes, together with vimentin and nestin. In addition to playing a structural role in astrocytes, where together with microtubules and microfilaments they form the cytoskeleton, these filaments are also involved in signal transmission. GFAP is also present in other central nervous system (CNS) cells, such as ependymal cells, in non-myelin-producing Schwann cells of the peripheral nervous system, and in enteric glia.
The protein is encoded by a single gene located on 17q21, which contains nine exons. At least 10 isoforms result from alternative splicing of GFAP pre-mRNA and the polyadenylation signal.14–17 GFAP-α (isoform 1) is the predominant isoform in the brain and spinal cord, but it also appears in the peripheral nervous system; it contains the classic 432 residues with full usage of the 9 exons of the GFAP gene. GFAP-δ or GFAP-¿ (isoform 2) is preferentially expressed by astrocytes from neurogenic niches including the subventricular zone and the hippocampus. GFAP-δ includes the use of an intron before exon 8 and has an alternative C-terminus and 431 residues. GFAP-δ is expressed in reactive astrocytes in diseases such as epilepsy, Alzheimer disease, and gliomas. The remaining isoforms are less frequent, although the association between some of these variants and neurodegenerative diseases has made them a subject of considerable research interest.18,19
Myelin is produced by oligodendrocytes in a dynamic process requiring 3 conditions: the presence of oligodendrocyte precursor cells (OPCs) in the demyelinated area, changes in oligodendrocyte form and membranes, and a favourable microenvironment. OPCs are immature oligodendrocytes that remain in the adult brain after embryonic development. They account for 5%-8% of the population of CNS glial cells20 and contribute to restoration of the myelin sheath, differentiating throughout adulthood. OPCs can express proteins including Olig2 and NG2. Myelin proteins produced and accumulated as a result of demyelination prevent remyelination through the protein kinase C α, Nogo 1, or LINGO1 signalling pathways.21,22 Semaphorins also play a role in regulating remyelination.23 Consequently, demyelination in patients with AxD may be explained by mechanisms with at least 4 effects24: (1) OPCs not being generated or surviving; (2) absence of stimuli promoting oligodendrocyte maturation; (3) prevention of myelin production by local inhibitory factors; or (4) presence of axonal alterations preventing myelination. This review analyses the hypotheses explaining myelin changes in patients with AxD.
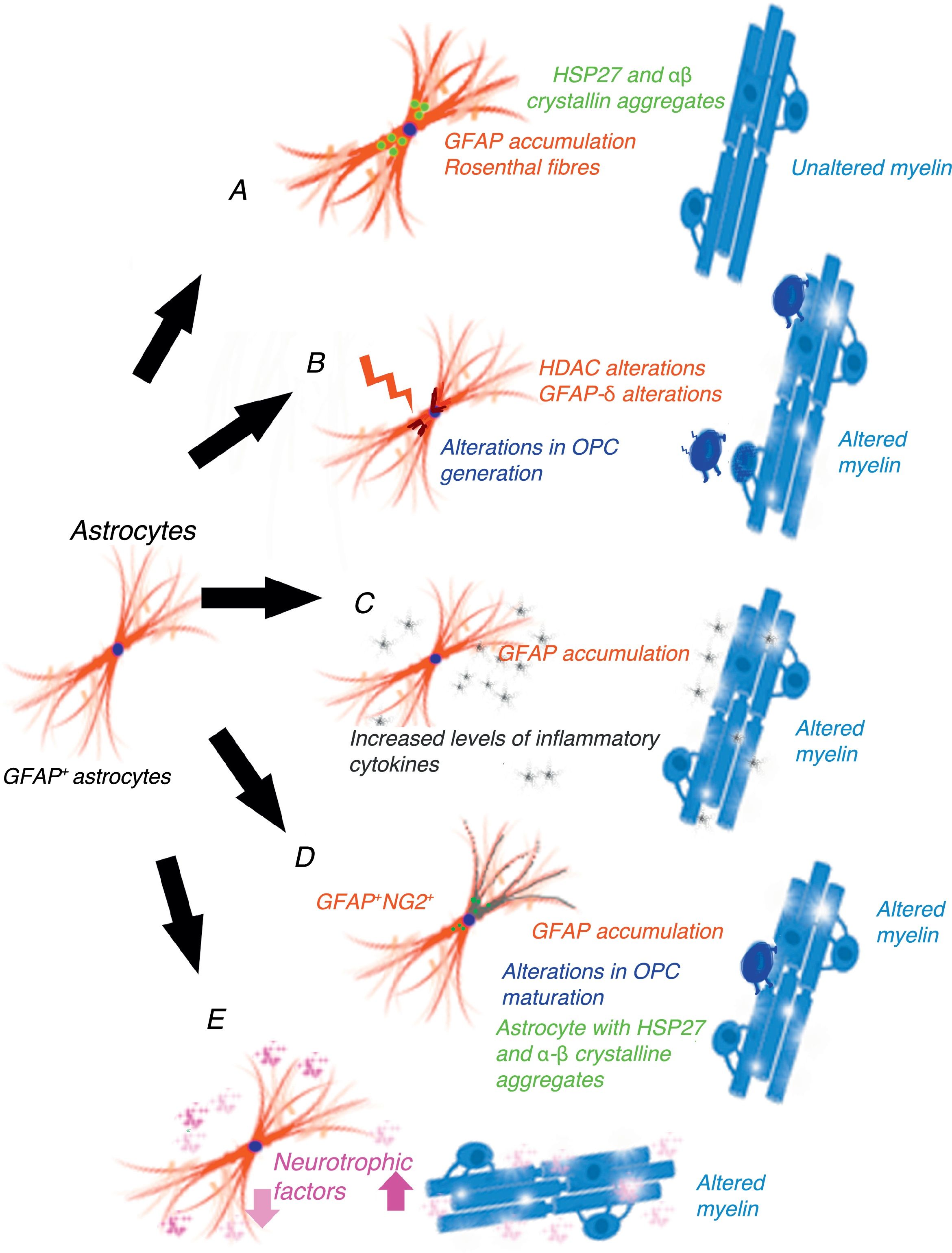
Gain-of-function mechanism in the glial fibrillary acidic proteinCho and Messing25 suggest that AxD is due to GFAP accumulation in astrocytes and that high levels of both mutant and wild-type GFAP are damaging. They propose a gain-of-function mechanism similar to that proposed for other neurodegenerative diseases, for example, in SOD1-dependent forms of amyotrophic lateral sclerosis.26 This mechanism is controversial, however.27 GFAP aggregates sequester HSP27, cathepsin, and α–βcrystallin; subsequent phosphorylation and ubiquitination generates Rosenthal fibres, triggering astrocyte damage28 and the activation of stress response pathways, such as the JNK and p38 pathways, in these astrocytes.29–31 Decreased GFAP degradation may be associated with decreased proteasome activity.32 Given GFAP's long half-life in vivo, interfering with protein degradation may have a prolonged effect, contributing to GFAP accumulation.33 This hypothesis has generated a considerable number of experiments and models, especially in mice with hyperexpression of mutant or wild-type GFAP. Tanaka et al.34 have published a study on transgenic mice with an R239H mutation. Messing and colleagues have studied 2 mouse models reproducing Rosenthal fibres and GFAP accumulation. These models provide valuable data on the mechanisms triggered by GFAP accumulation.35–40 Other researchers have attempted to establish a correlation between disease prognosis and CSF GFAP level.41 The main weakness of this hypothesis is that this mechanism does not cause demyelination42 (Fig. 1A). A further limitation is the fact that gliomas displaying GFAP accumulation and Rosenthal fibres do not show myelin loss; it is therefore reasonable to question the assertion that increased GFAP levels alone may explain the disease.
Epigenetic alterations to transcription Gain-of-function mechanism. (B) Epigenetic alterations on transcription. (C) The inflammatory mechanism. (D) GFAP+/NG2+ cells. (E) Post-transcriptional alterations of GFAP.")
GFAP expression levels are regulated by the activity of the GFAP gene promoter; this process is largely dependent on epigenetic alterations. During astrogenesis in neural stem cells, GFAP promoter demethylation activates GFAP transcription43–46; histone acetylation controls GFAP expression during neural stem-cell differentiation and depends on the cell's stage of differentiation.47,48 The degree of histone acetylation is regulated by the enzyme's histone acetylase and histone deacetylase (HDAC). Kanski et al.49 have shown that histone acetylation in astrocytes is an important regulator of the transcription and alternative splicing of GFAP. HDAC inhibition significantly reduces GFAP expression in primary human astrocytes and in astrocytoma cells. This mechanism is noteworthy given that HDAC inhibition modifies the balance between alternative transcription of GFAP-δ and the constitutive isoform GFAP-α, favouring GFAP-δ expression, which is modified during differentiation into astrocyte or oligodendrocyte lineage cells. Inhibition of HDAC activity modifies the structure of intracellular GFAP filaments in terms of length, location, and degree of aggregation. Several studies have found an association between changes in GFAP aggregation and alterations in differentiation in patients with leukodystrophies,50–52 associated with differences in GFAP-δ expression. The literature includes a report of a patient with mutations in GFAP and HDAC6 who had a severe phenotype of AxD53 and showed reduced HDAC6 activity. According to these authors’ hypothesis, myelin loss in AxD is explained by GFAP aggregation and alterations in oligodendrocyte differentiation (Fig. 1B).
The inflammatory mechanismOlabarria et al.54 suggest that AxD may be mediated by an inflammatory mechanism. The demyelination observed in multiple sclerosis and neuromyelitis optica is of autoimmune origin and partly attributable to inflammatory mechanisms and increased levels of proinflammatory cytokines. Kondo et al.55 studied induced pluripotent stem cells (iPSCs) from 3 patients with AxD and different GFAP mutations. AxD iPSC-derived astrocytes showed GFAP+ cytoplasmic aggregates, such as Rosenthal fibres, and altered cytokine release. Some authors report moderate lymphocytic infiltration and microglial activation in the brains of patients with AxD.56,57 These authors have studied cytokine expression in mouse models with GFAP overexpression and mice heterozygous for the GFAP-R236H mutation, detecting an inflammatory response. The iPSCs taken from the patients with AxD showed increased levels of proinflammatory cytokines such as GM-CSF, IL-5, IL-6, and tumour necrosis factor-α. It has also been reported that the GFAP molecule may be deaminated,58 which may trigger an autoimmune response.59 In light of the above, these authors suggest that a neuroinflammatory process may be involved in AxD pathogenesis and that myelin loss may be due to an autoimmune mechanism, as occurs with multiple sclerosis (Fig. 1C).
GFAP+/NG2+ cellsNG2+ glial cells,60,61 also known as synantocytes62 or polydendrocytes,63 represent 8%-9% of all white matter cells and 2%-3% of grey matter cells.64 They were initially identified as Olig2 progenitor cells65 as they differentiated into oligodendrocytes.66–70 NG2+ glial cells are in fact OPCs capable of receiving synaptic input, therefore regulating their own differentiation. These cells can also extend their processes to the node of Ranvier and transform into reactive astrocytes in pathological conditions and in cell cultures71,72; they therefore represent a transitional stage between olygodendrocytes and astrocytes.73 NG2+ cells may be GFAP+ in certain pathological conditions or locations, such as the area surrounding the demyelination in multiple sclerosis.74 Oligodendrocyte lineage cells have also been found to be GFAP+ in patients with leukodystrophies and autoimmune or virus-induced myelin alterations.75–77 Mice with overexpression of mutant GFAP display altered hippocampal neurogenesis.78 Our research group observed the same findings in a cell model transfected with AxD mutations.79 GFAP expression makes Schwann cells behave functionally as astrocytes, not generating myelin.80 GFAP expression has been observed in oligodendroglial lineage cells in the initial stages; NG2 expression has been observed in cells that will subsequently differentiate into astrocytes81–84; copresence of GFAP and NG2 expression makes it very difficult to determine whether cells are oligodendrocytes or astrocytes. OPC transplantation into the CNS generates oligodendrocytes and astrocytes simultaneously.85 Persistence of GFAP expression in cells that should differentiate into oligodendrocyte lineage cells would explain GFAP accumulation and the absence of myelin production (Fig. 1D). Persistence of GFAP expression in NG2+ cells, and consequently differentiation into a specific lineage, is very likely to depend on the GFAP isoform or on factors related to the microenvironment where maturation occurs.
Post-transcriptional alterations of GFAPAstrocytes secrete high levels of neurotrophic factors,86 some of which are involved in myelination. Some experimental models have shown that BDNF is involved in oligodendrocyte proliferation and in remyelination.87,88 The interaction between astrocytes and oligodendrocytes may play a crucial role in myelination.89,90 Experiments with cell cultures provide direct evidence that BDNF in astrocytes promotes OPC maturation; in vivo studies have observed reduced oligodendrogenesis in transgenic mice showing reduced astrocytic BDNF regulation. Other growth factors, including CNTF,91 FGF, TGF, and GDNF, as well as nuclear receptors, may modify the activation of GFAP transcription.92 Yang and Wang93 extensively reviewed the complex mechanisms occurring after GFAP transcription. GFAP undergoes a series of post-translational changes and is highly regulated by protein kinases; some genetic and environmental changes affecting the molecule may have an impact on astrocyte function, altering myelination (Fig. 1E).
ConclusionThe hypothesis of the gain-of-function mechanism suggests the possibility that we may discover a drug that could improve patients’ condition by reducing GFAP expression. Great efforts have been made to develop effective treatments with cell or mouse models of GFAP hyperexpression.94,95 However, if the changes induced by GFAP hyperexpression are secondary, such treatments would be symptomatic, and therefore insufficient for a disease starting during embryonic development or the neonatal period.
The different hypotheses discussed may explain myelin alterations in patients with AxD; some of these mechanisms may co-occur. Administering treatments capable of modifying the epigenetic mechanisms of the disease96 is a reasonable and entirely valid option; this line of treatment is already being evaluated in clinical trials in the context of cancer.97 Likewise, the differentiation of immature cells into astrocyte and oligodendrocyte lineage cells may constitute another therapeutic target.
Conflicts of interestThe authors have no financial or commercial relationships that could create conflicts of interest with regard to this article.
We would like to thank the Ayuda Juanma Foundation (www.ayudajuanma.es) for funding research into AxD at Hospital Clínico San Carlos.
Please cite this article as: Gómez-Pinedo U, Duran-Moreno M, Sirerol-Piquer S, Matias-Guiu J. La alteración de la mielina en la enfermedad de Alexander. Neurología. 2018;33:526–533.
