




El retraso global del desarrollo (RGD) y la discapacidad intelectual (DI) son motivos de consulta frecuentes en la práctica neuropediátrica. El rendimiento de los estudios diagnósticos en niños con RGD/DI varía ampliamente y, en consecuencia, no hay acuerdo universal respecto a los estudios que se deben realizar.
Material y métodoRevisamos nuestra experiencia en el diagnóstico etiológico de los niños con RGD/DI valorados en la consulta de Neuropediatría durante un periodo de 5 años: 2006-2010.
ResultadosDurante el periodo de estudio fueron valorados 995 niños con RGD/DI. El diagnóstico etiológico fue establecido en 309 (31%) y no en 686 (69%), a pesar de múltiples estudios realizados. En 142 niños, el 46% de los casos con diagnóstico etiológico establecido, la causa es genética: 118 encefalopatías genéticas y 24 enfermedades metabólicas hereditarias. Nuestros datos indican que establecer un diagnóstico etiológico es más fácil cuando el RGD/DI está asociado a parálisis cerebral infantil, epilepsia, espasmos infantiles/síndrome de West o déficit visual, pero más difícil en casos de trastorno del espectro autista. Los estudios genéticos están incrementando los diagnósticos etiológicos y constituyéndose en el primer escalón de estudio. El microarray comparative genomic hybridisation es la prueba con mayor rentabilidad diagnóstica en el estudio de RGD/DI.
DiscusiónEl coste-efectividad de los exámenes complementarios es aparentemente bajo en ausencia de orientación clínica. Incluso en ausencia de tratamiento, el diagnóstico etiológico es importante para establecer un consejo genético y posible diagnóstico prenatal, resolver cuestiones a padres y profesionales, y cesar la realización de más pruebas complementarias.
Global developmental delay (GDD) and intellectual disability (ID) are common reasons for consultation in paediatric neurology. Results from aetiological evaluations of children with GDD/ID vary greatly, and consequently, there is no universal consensus regarding which studies should be performed.
Material and methodWe review our experience with determining aetiological diagnoses for children with GDD/ID who were monitored by the paediatric neurology unit over the 5-year period between 2006 and 2010.
ResultsDuring the study period, 995 children with GDD/ID were monitored. An aetiological diagnosis was established for 309 patients (31%), but not in 686 (69%), despite completing numerous tests. A genetic cause was identified in 142 cases (46% of the total aetiologies established), broken down as 118 cases of genetic encephalopathy and 24 of metabolic hereditary diseases. Our data seem to indicate that diagnosis is easier when GDD/ID is associated with cerebral palsy, epilepsy, infantile spasms/West syndrome, or visual deficit, but more difficult in cases of autism spectrum disorders. Genetic studies provide an increasing number of aetiological diagnoses, and they are also becoming the first step in diagnostic studies. Array CGH (microarray-based comparative genomic hybridisation) is the genetic test with the highest diagnostic yield in children with unexplained GDD/ID.
DiscussionThe cost-effectiveness of complementary studies seems to be low if there are no clinically suspected entities. However, even in the absence of treatment, aetiological diagnosis is always important in order to provide genetic counselling and possible prenatal diagnosis, resolve family (and doctors’) queries, and halt further diagnostic studies.
El retraso psicomotor global (RGD) y la discapacidad intelectual (DI) son motivos de consulta frecuentes en la práctica neuropediátrica. La prevalencia estimada de retraso psicomotor y retardo mental oscila entre un 1 y un 10%1. Entre un 50 y un 80% de los casos no tienen un diagnóstico etiológico establecido2. El rendimiento de la evaluación diagnóstica de niños con RGD/DI varía ampliamente (10-81%), reflejando muchos factores, incluidas diferencias de las poblaciones estudiadas y extensión de los estudios realizados, lo que dificulta las revisiones sistemáticas3. En consecuencia, no hay acuerdo universal respecto a los estudios que deben ser realizados ante los niños afectados de RGD/DI1-11. Además, los estudios deben actualizarse y adaptarse a los continuos avances sociales, técnicos y científicos. Se revisa de forma retrospectiva la experiencia de la sección de Neuropediatría en el diagnóstico etiológico en los casos afectados de RGD/DI en un periodo de 5 años, entre 2006 y 2010.
Material y métodoLa unidad de Neuropediatría de nuestro hospital (hospital terciario) fue creada en 1990 y dispone de una base de datos con todos los casos valorados en la unidad desde su inicio en mayo de 1990. Dicha base de datos es actualizada cada vez que se produce una novedad clínica, evolutiva, de tratamiento o de exámenes complementarios12-14.
En 2008, ampliamos nuestra estrategia diagnóstica ante diferentes problemas neuropediátricos, como encefalopatías prenatales o RGD/DI, en un intento de identificar alteraciones genéticas y enfermedades metabólicas hereditarias (EMH). Aplicamos dicha estrategia a casos nuevos y casos antiguos sin diagnóstico etiológico. A la mayoría de los niños incluidos en la revisión se les ha hecho un extenso estudio bioquímico en sangre y orina, neuroimagen (en la mayoría resonancia magnética (RM) cerebral), cariotipo y estudio de síndrome X-frágil y otros estudios genéticos orientados; dichos estudios quedan recogidos en resultados. Diagnosticamos de RGD a aquellos pacientes que, con una edad inferior a 5 años, presentaban una alteración del neurodesarrollo en 2 o más áreas según la escala de desarrollo de Denver o Llevant. Diagnosticamos de DI a aquellos niños que, con una edad igual o mayor a 5 años, disponían de un estudio de cociente intelectual < 85 o bien precisaban educación especial o adaptación curricular significativa.
Revisamos nuestra experiencia en el diagnóstico etiológico en los casos afectados de RGD/DI en un periodo de 5 años, entre 2006 y 2010.
Este es un estudio de incidencia de casos en este periodo y de prevalencia de aquellos casos diagnosticados previamente y controlados durante el periodo de estudio. Se incluye a niños con RGD/DI aislado o asociado a otras afecciones concomitantes, como parálisis cerebral infantil (PCI), trastorno del espectro autista (TEA) o epilepsia; se incluyen también casos de regresión. Se excluyen TEA con inteligencia normal o de altas capacidades. Se revisan los diagnósticos y los estudios realizados en los casos sin diagnóstico etiológico establecido.
Se ha realizado una prueba de chi cuadrado de Pearson con un grado de libertad para establecer la asociación estadística entre las diferentes afecciones concomitantes y padecer o no RGD/DI.
ResultadosEntre el 1 de enero del 2006 y el 31 de diciembre del 2010, la base de datos fue actualizada en 6.018 niños. De estos, un total de 995 (16,5%) presentaban RGD/DI.
En 686 casos (69%), a pesar de ser realizados muchos estudios, no se alcanzó un diagnóstico etiológico y sí se alcanzó en 309 casos (31%) (tabla 1).
Casos con diagnóstico etiológico entre pacientes con retraso global del desarrollo y discapacidad intelectual (n=309)
| Enfermedades | N |
| Encefalopatías prenatales disruptivas | 41 (13%) |
| Infecciones congénitas (13 CMV, 3 toxoplasmosis) | 19 |
| Encefalopatías teratogénicas (16 SAF) | 18 |
| Encefalopatías disruptivas gemelares | 4 |
| Encefalopatías genéticas | 118 (38%) |
| Síndrome de Down | 12 |
| Síndrome X-frágil | 7 |
| Deleciones subteloméricas | 7 |
| Síndrome de Patau | 1 |
| Otras cromosomopatías diagnosticadas por array CGHa | 21 |
| Complejo esclerosis tuberosa | 11 |
| Distrofia miotónica congénita | 4 |
| Síndrome de Rett (4 MECP2 y 2 CDKL5 mutaciones) | 6 |
| Espectro Dravet con mutaciones en SCN1A | 9 |
| Síndrome Prader Willi | 5 |
| Síndrome Angelman | 4 |
| Neurofibromatosis tipo 1 | 5 |
| Otras encefalopatías genéticasb | 16 |
| Errores congénitos del metabolismo | 24 (7,8%) |
| Enfermedades mitocondriales | 3 |
| Enfermedades lisosomalesc | 7 |
| Enfermedades del metabolismo intermediariod | 6 |
| X-ALDe | 3 |
| Otros ECMf | 5 |
| Encefalopatías perinatales | 102 (33%) |
| Encefalopatías posnatalesg | 20 (6,55%) |
| Tumores cerebrales | 4 (1,3%) |
| Tumor hemisférico | 2 |
| Tumor cerebeloso | 1 |
| Tumor de línea media | 1 |
Array CGH: microarray comparative genomic hybridisation; CMV: citomegalovirus; SAF: síndrome alcohólico fetal; X-ALD = adrenoleucodistrofia ligada a X.
Otras encefalopatías genéticas: 3 lisencefalias con mutación LIS1, 3 distrofinopatías, 2 síndromes Williams Beuren, 2 síndromes de Joubert, un síndrome CRASH con mutación L1CAM, una sintelencefalia con deleción ZIC2, un síndrome Proteus, un Waardenburg, un Cofin Siris y un Cornelia de Lange.
Enfermedades lisosomales: un Hurler, un NCL2 ceroidolipofuscinosis, un Krabbe y 4 leucodistrofias metacromáticas.
Seis enfermedades del metabolismo intermediario: 2 hermanos con aciduria hidroxibutírica, una homocistinuria, una hiperglucinemia no cetósica, una enfermedad de orina con olor de jarabe de Arce y una deficiencia de biotinidasa.
De entre los casos en los que se estableció el diagnóstico etiológico, 142 (46%) están genéticamente determinados: 118 encefalopatías genéticas y 24 EMH, y en 126 el diagnóstico se realizó sobre la base de la historia clínica (102 encefalopatías perinatales, 20 encefalopatías posnatales y 4 tumores cerebrales). Estos casos diagnosticados mediante la historia clínica se han excluido del estudio analítico. De esta forma, el total de casos analizados con RGD/DI son 869 (686 sin diagnóstico establecido y 183 con diagnóstico).
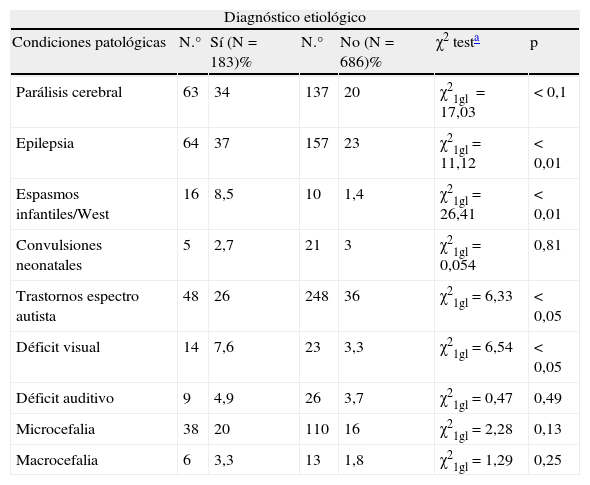
Hemos analizado la relación entre obtener un diagnóstico etiológico y la asociación con una condición particular como PCI, epilepsia, espasmos infantiles/síndrome de West, microcefalia, macrocefalia, convulsiones neonatales, TEA, déficit visual o hipoacusia en el total de casos con RGD/DI, excluyendo a aquellos (n=126) en los que el diagnóstico se estableció sobre la base de la historia clínica. Nuestros datos muestran una asociación estadísticamente significativa entre obtener un diagnóstico etiológico y presentar PCI, epilepsia, espasmos infantiles/síndrome de West e hipovisión. No hay una asociación estadísticamente significativa entre obtener un diagnóstico etiológico y TEA (tabla 2).
Diagnóstico etiológico en pacientes con RGD/DI y otras afecciones, y la relación entre obtener un diagnóstico etiológico y presentar las distintas afecciones. Los casos debidos a encefalopatías prenatales y perinatales y tumores cerebrales se han excluido
| Diagnóstico etiológico | ||||||
| Condiciones patológicas | N.° | Sí (N=183)% | N.° | No (N=686)% | χ2 testa | p |
| Parálisis cerebral | 63 | 34 | 137 | 20 | χ21gl=17,03 | < 0,1 |
| Epilepsia | 64 | 37 | 157 | 23 | χ21gl=11,12 | < 0,01 |
| Espasmos infantiles/West | 16 | 8,5 | 10 | 1,4 | χ21gl=26,41 | < 0,01 |
| Convulsiones neonatales | 5 | 2,7 | 21 | 3 | χ21gl=0,054 | 0,81 |
| Trastornos espectro autista | 48 | 26 | 248 | 36 | χ21gl=6,33 | < 0,05 |
| Déficit visual | 14 | 7,6 | 23 | 3,3 | χ21gl=6,54 | < 0,05 |
| Déficit auditivo | 9 | 4,9 | 26 | 3,7 | χ21gl=0,47 | 0,49 |
| Microcefalia | 38 | 20 | 110 | 16 | χ21gl=2,28 | 0,13 |
| Macrocefalia | 6 | 3,3 | 13 | 1,8 | χ21gl=1,29 | 0,25 |
N.°: número de casos con una condición particular; RGD/DI: retraso global del desarrollo y discapacidad intelectual; p: asociación estadísticamente significativa entre presentar una condición particular y la posibilidad de obtener un diagnóstico etiológico; %: porcentaje de pacientes en cada condición con o sin diagnóstico etiológico respecto del total de pacientes.
En el grupo de RGD/DI sin diagnóstico etiológico establecido, se realizaron los siguientes exámenes complementarios:
- –
Estudios bioquímicos normales: 550 amonios, 518 aminoácidos, 525 lácticos, 497 CK, 448 hormonas tiroideas, 340 ácidos grasos de cadena larga, 282 test de toluidina, 298 homocisteínas, 288 ceruloplasminas, 281 cobres, 250 ácidos orgánicos, 179 test de CDT (detección de síndrome defectos congénitos de la glucosilación de las proteínas [CDG]) y 13 estudios de neurotransmisores en el líquido cefalorraquídeo.
- –
Estudios genéticos normales: 457 cariotipos, 399 síndrome X-frágil, 59 deleciones subteloméricas, 50 array CGH, 46 síndrome de Angelman, 27 enfermedad de Prader Willi, 18 de distrofia miotónica, 12 mutaciones MECP2, 7 CDKL5, CDKL5 16 de SCN1A y GABRG2. Realizamos estudio retrospectivo de ADN de citomegalovirus en sangre seca procedente de prueba cribado neonatal en 9 casos.
- –
Estudios normales de RM cerebral en 284 casos, de TAC craneal en 141 y de RM espectroscópica en 69.
- –
Estudios de neuroimagen alterados, aunque no establecieron diagnóstico etiológico: 107 RM y 77 TAC craneales.
En nuestra serie, es más fácil establecer un diagnóstico etiológico cuando el RGD/ID se asocia a PCI, espasmos infantiles/síndrome de West o déficit visual, pero más difícil en casos de TEA (tabla 2).
Obviamente, hay casos de fácil diagnóstico sobre la base de la clínica, como el síndrome de Down, la esclerosis tuberosa o la neurofibromatosis 1, que incrementan la rentabilidad diagnóstica de los estudios complementarios, especialmente genéticos.
No hemos analizado la existencia de rasgos dismórficos o manchas en la piel, que aumentan la posibilidad de obtener un diagnóstico.
No ha sido posible relacionar el grado de discapacidad intelectual con la posibilidad de llegar a un diagnóstico etiológico; en casos leves, el diagnóstico etiológico es más difícil de obtener.
El estudio de EMH en niños con RGD/DI tiene una rentabilidad diagnóstica que oscila entre un 0,2 y un 4,6%, dependiendo de la presencia de indicadores clínicos y la amplitud de los estudios realizados. El test de CDT (% de transferrina deficientemente carboxilada) para el diagnóstico de los síndromes CDG tiene una rentabilidad de hasta un 1,4% y el estudio de los defectos de síntesis y transporte de creatina de hasta un 2,8%8. En nuestra serie, no se han realizado estudios en orina de defectos de síntesis y transporte de creatinina; aunque estos defectos también pueden ser detectados con la RM espectroscópica, deben ser incluidos en los estudios sistemáticos, dado que existe opción de tratamiento.
Estudios bioquímicos no orientados tienen muy poca rentabilidad y tienen poco beneficio en niños mayores de 2-3 años con aislado RGD/DI.
Encefalopatías genéticas, infecciones congénitas, enfermedades peroxisomales, mitocondriales, lisosomales, síndromes CDG y otras EMH pueden ser indistinguibles en etapas precoces sobre la base de la clínica. Las EMH son una causa rara de RGD/DI (aproximadamente 1%), particularmente si no hay signos o síntomas que indiquen un problema metabólico. Sin embargo, el efecto de identificar una EMH en el pronóstico del paciente puede ser sustancial. Puede ser que estudiar relativamente raras EMH tenga más impacto sobre las familias y la sociedad que estudiar síndromes genéticos, dado que pueden influir en el tratamiento y pronóstico. La posibilidad de instaurar un tratamiento efectivo debe tener una influencia en la práctica clínica, por encima de exclusivamente la rentabilidad diagnóstica3,8.
No existe consenso universal respecto a la indicación de neuroimagen en la evaluación del niño con RGD/DI. Las recomendaciones varían desde la realización de neuroimagen a todos hasta limitarlo solo a casos de indicaciones clínicas3. Actualmente, salvo en ciertas emergencias, la prueba de elección en el estudio de cualquier encefalopatía es la RM. La neuroimagen establece el diagnóstico etiológico en algunos casos, como en nuestros 2 casos de síndrome de Joubert y en algunos casos de esclerosis tuberosa. Aunque en algunos casos no establece el diagnóstico, puede ayudar a identificar la causa de un caso particular, de la misma manera que el examen dismorfológico puede llevar al diagnóstico clínico3. La RM guió el diagnóstico etiológico en nuestros casos de lisencefalias, síndrome CRASH, sintelencefalia, infección congénita por citomegalovirus y leucodistrofias.
Los estudios genéticos están aumentando su rentabilidad diagnóstica y constituyéndose en el primer paso de estudio. En niños con RGD/DI, el array CGH es diagnóstico en un 7,8% de casos y en un 10,6% en aquellos con rasgos sindrómicos. El cariotipo de alta resolución es anormal en al menos un 4,6% y en un 18,6% en casos de rasgos sindrómicos. El estudio FMR1 de síndrome de X-frágil es patológico en al menos el 2% de los pacientes con leve o moderado RGD/DI. El estudio del gen MECP2 es diagnóstico en el 1,5% de las niñas con moderado o severo RGD/DI y en menos del 0,5% de los varones con RGD/DI8.
El array CGH es la prueba genética con mayor rentabilidad diagnóstica en niños con RGD/DI de causa no establecida8. Ha establecido el diagnóstico en el 29,5% de nuestros casos (21 de 71).
Las nuevas generaciones de paneles de secuenciación masiva seguramente revolucionarán la estrategia diagnóstica del estudio etiológico de la discapacidad intelectual15.
El coste-efectividad de los estudios complementarios es aparentemente bajo en ausencia de orientación clínica. La cuestión es si hacemos demasiados estudios, aunque debemos identificar problemas mayores, como el síndrome X-frágil: 15 casos identificados de entre 468 estudiados. La identificación precoz de otros trastornos de transmisión vertical, como la distrofia miotónica o la distrofia muscular de Duchenne, es una gran responsabilidad desde el punto de vista diagnóstico.
En el proceso diagnóstico también debería considerarse la ausencia de anomalías (hallazgos negativos)5.
Este es un estudio retrospectivo que muestra nuestra estrategia diagnóstica hasta recientemente. Creemos en la importancia de analizar nuestro trabajo y compartir nuestra experiencia, aunque admitimos la heterogeneidad de nuestra población, con casos de fácil diagnóstico y otros de regresión, pero pretendíamos evitar un sesgo de selección.
El RGD/DI de causa no establecida está en la mayoría de los casos genéticamente determinado y tiene un alto impacto social, familiar y económico, lo que lleva a una elevada demanda de diagnóstico precoz. La mayoría de los casos de RGD/DI son enfermedades raras y la Comunidad Europea recomienda a sus estados miembros que establezcan un plan nacional de enfermedades raras16; en España, la segunda línea estratégica de este Plan Nacional es la prevención y detección precoz17.
Tras esta revisión, hemos decidido cambiar nuestra estrategia de estudio, resaltando que para establecer diagnósticos, además de las estrategias, es necesario el control evolutivo y, en muchos casos, un planteamiento individualizado.
En niños mayores de 2-3 años con aislado RGD/DI, con o sin TEA, siempre que no haya cambios evolutivos, hemos decidido realizar solo una analítica elemental con enzimas musculares en varones (para identificar casos de distrofia muscular de Duchenne), homocisteína, test de CDT y hormonas tiroideas. En menores de 2-3 años, en los que pueden esperarse cambios evolutivos, seguiremos realizando un amplio estudio neurometabólico tratando de identificar precozmente la EMH.
El estudio genético, con cariotipo de alta resolución y estudio de X-frágil, lo realizaremos a todo RGD/DI sin diagnóstico, salvo el estudio de X-frágil a niñas con microcefalia evidente o polimalformadas. Realizaremos array CGH ante la presencia de determinados factores, como antecedentes familiares de DI o de abortos, retraso de crecimiento intrauterino, micro o macrocefalia, micro o macrosomía, rasgos dismórficos o malformaciones asociadas cardíacas, renales, oftalmológicas u otras.
La RM cerebral la realizaremos ante epilepsia, focalidad neurológica, hipoacusia, déficit visual o alteraciones oftalmológicas, macrocefalia o franca aceleración del crecimiento cefálico y microcefalia, o franco estancamiento del crecimiento cefálico.
Para concluir, queremos enfatizar la importancia del diagnóstico en las enfermedades raras tratables. Sin embargo, incluso en ausencia de tratamiento, el diagnóstico etiológico es siempre importante para establecer el riesgo de repetición, el consejo genético y el posible diagnóstico prenatal, resolver las preguntas e incertidumbres de familiares y profesionales, y cesar los estudios diagnósticos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

