



Posterior reversible encephalopathy syndrome (PRES) is a clinical and radiological entity characterised by the acute or subacute onset of headache, altered level of consciousness, visual alterations, seizures, nausea, and vomiting; it also causes neuroimaging alterations, which are generalised, reversible, and predominantly posterior.1–3 It usually manifests in the context of systemic diseases; in children, it has been identified in the context of kidney failure, immunosuppressant treatment and chemotherapy, such autoimmune diseases as systemic lupus erythematosus, and idiopathic arterial hypertension (AHT), among others.1,4,5 Approximately 70%-80% of patients present moderate to severe AHT.2,6,7 Brain magnetic resonance imaging (MRI) is essential for diagnosis as it identifies the presence of oedema surrounding the white matter bilaterally, mainly in the posterior area (parietal and occipital lobes).2,3,8,9 The pathophysiology of PRES is unknown; several mechanisms have been suggested, and probably coexist in some cases: loss of autoregulatory vascular tone causing hyperperfusion, systemic vasoconstriction with hypoperfusion, and dysfunction or endothelial injury with lesion to the blood-brain barrier.9,10 Symptoms fully resolve when the underlying cause is corrected early; otherwise, however, the condition may result in such irreversible damage as cortical blindness or death. MRI abnormalities disappear in follow-up examinations performed after the proper treatment is administered.11–13
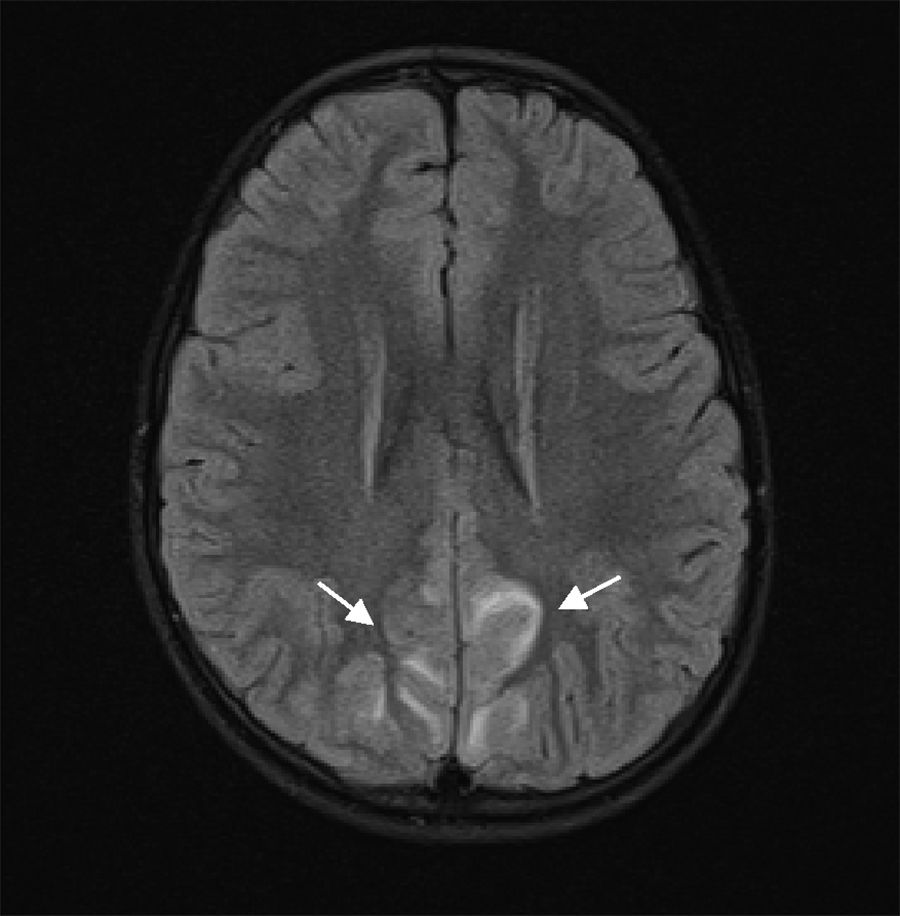
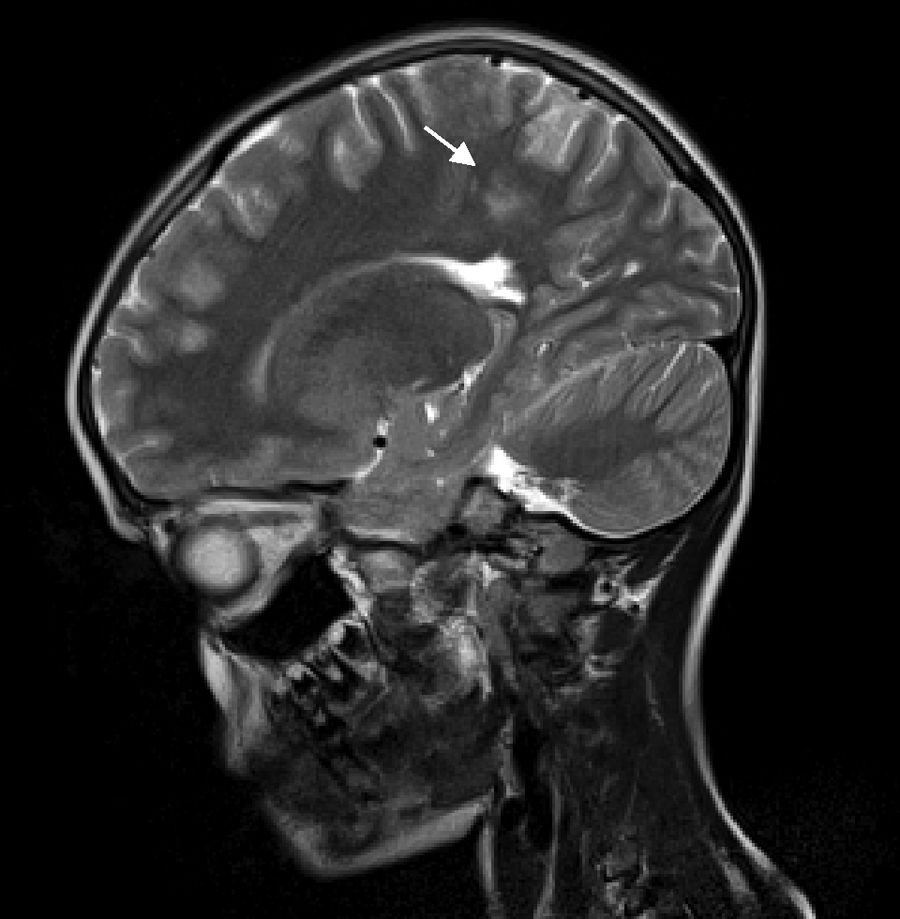
Our patient is a 6-year-old girl with no relevant personal history, who attended the emergency department due to a 4-day history of headache, vomiting, abdominal pain, and lack of mobility. During the examination, the patient was drowsy but easily awakened, with the neurological examination detecting no other alterations (eye fundus with no papilloedema). During the first hour after admission, she presented weak mastication and empty gaze, and blood pressure values were observed to be persistently above the 95th percentile. A head computed tomography scan revealed cortico-subcortical hypodensities in the left anterior parietal and parasagittal regions, and an electroencephalography showed a slow, poorly reactive, and ill-defined rhythm, suggesting cortico-subcortical involvement. A brain MRI scan showed multifocal, T2-hyperintense lesions symmetrically surrounding the white matter of the temporo-parietal and occipital regions and the left frontal lobe, suggesting PRES (Figs. 1 and 2). Other such diagnostic hypotheses as stroke, cerebral venous thrombosis, central nervous system vasculitis, and encephalitis were ruled out. For the aetiological diagnosis of AHT and its consequences, we requested the following studies: urine sediment analysis, kidney function test, serum electrolyte study, lipid profile, immunology study, 24-hour urine catecholamines, renal ultrasound, an echo-Doppler study of the renal arteries, and an echocardiogram; all tests yielded normal results without identifying any secondary cause of AHT.
.")

Blood pressure was controlled on day 9 of hospitalisation using enalapril and hydrochlorothiazide plus amiloride; subsequent clinical progress was favourable (with no de novo symptoms). The imaging study performed 9 days after treatment onset revealed almost complete recovery of the lesions diagnosed by MRI. The patient was discharged on day 12 and referred to the neuropaediatric outpatient consultation. Antihypertensives were suspended after 8 weeks of treatment; 12 weeks after the initial episode, the patient presented normal blood pressure, with a brain MRI scan showing complete resolution of the lesions.
PRES is a rare syndrome in children; however, given the low level of suspicion, its incidence may be underestimated.13 This case presents atypical manifestations, since the symptoms appeared in a previously healthy girl in the context of sudden-onset AHT, with no identified aetiology and with symptoms resolving in 8 weeks.
Although the majority of the cases described in the literature are associated with systemic diseases,6,14 this syndrome has been reported in association with idiopathic AHT.2 Favourable clinical progression was observed after blood pressure control, with complete resolution of the lesions after 12 weeks. Furthermore, the AHT episode resolved without aetiology being confirmed.
Most authors recommend performing a new imaging assessment after symptom resolution, although there is no consensus on the ideal time for the study since resolution is observed between 8 days and 17 months after the initial episode.1,14
The progression time necessary for lesions to become irreversible is not well determined.
With this case report, we hope to raise awareness of a reversible clinical and radiological entity and to highlight the importance of timely diagnosis and early treatment of the underlying cause to prevent permanent neurological sequelae.
recomendados

Neurología (English Edition) sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas