




We examine those prenatal encephalopathies with clinical or neuroimaging data of encephalopathy before the birth. They affect a significant number of children seen by paediatric neurologists. They can be of disruptive origin (due to vascular problems, drugs, toxins or congenital infections), and genetically determined. We include cases of autism spectrum disorder and mental retardation with no history of perinatal of postnatal damages.
Material and methodsWe analysed our 19-year neuro-paediatric data base in search of prenatal encephalopathies and their diagnostic origin. We also analyse the studies made in the cases with a diagnosis of unknown origin.
ResultsThe 19-year period of study in the data base included 11,910 children, and 1596 (13.5%) were considered as prenatal encephalopathies; 1307 children (81.4%) had a diagnosis of unknown origin, despite many investigations being done in a large number of them.
DiscussionMost of the children included in this study suffer a rare disease, and whether they are identified or not, they increasingly require an early diagnosis. Peroxisomal, mitochondrial, lysosomal diseases, carbohydrate glycosylation deficiency syndrome and other inborn error of metabolism, congenital infections and genetic encephalopathies, can be clinically indistinguishable in early life and require specific studies to identify them. Early diagnosis requires strategies using step-wise systematic studies, giving priority to those diseases that could be treated, and in many cases using an individualised approach. We believe that the potential benefits of early diagnosis, including savings on further studies, genetic counselling and prenatal diagnosis, overcome the financial costs.
Consideramos encefalopatías prenatales las que tienen datos clínicos o prenatales de encefalopatía antes del nacimiento. Afectan a un número importante de niños controlados en las consultas de neuropediatría. Pueden ser disruptivas (por problemas vasculares durante el embarazo, drogas, tóxicos o infecciones congénitas), y genéticamente determinadas. Incluimos casos de trastorno del espectro autista y retardo mental sin historia de sufrimiento perinatal o postnatal.
Material y métodosSe revisa nuestra experiencia en el diagnóstico etiológico de las encefalopatías prenatales durante los últimos 19 años. Se analizan los estudios realizados en los casos sin diagnóstico etiológico.
ResultadosEn el periodo de estudio de 19 años y 5 meses, en la base de datos de neuropediatría figuran 11.910 niños. Tienen establecido el diagnóstico de encefalopatía prenatal 1596 (13,5%). De ellos no tienen diagnóstico etiológico preciso 1.307 niños (81,4%) pese a habérseles realizado múltiples estudios complementarios, fundamentalmente bioquímicos, genéticos y de neuroimagen.
DiscusiónMuchos de los niños incluidos en este estudio presentan enfermedades raras, estén o no identificadas, que demandan crecientemente un diagnóstico precoz. Enfermedades peroxisomales, lisosomales, mitocondriales, defectos congénitos de la glucosilación, entre otras enfermedades metabólicas hereditarias, infecciones congénitas, cromosomopatías y genopatías, pueden ser indistinguibles clínicamente y necesitan estudios específicos para su identificación. Un diagnóstico precoz precisa estrategias de estudios sistemáticos de forma escalonada, priorizando las enfermedades que tienen posibilidades terapéuticas y en muchos casos es necesaria también una aproximación individualizada. Creemos que las ventajas potenciales del diagnóstico precoz, incluido el ahorro de más pruebas, y la prevención, probablemente sobrepasan el gasto financiero.
The most frequent health care process that neuro-paediatric professionals are involved in begins with the motive for the first consultation, from which a diagnosis that can have up to four different levels is sought:
- 1.
Topographic location of the problem: encephalopathy (which, in turn, may be exclusive or with associated involvement of the cerebral hemispheres, cerebellum, midline, etc.); we must always consider the possible involvement of the visual and auditory pathways, myelopathy, disorder of the neuromuscular unit (anterior horn, peripheral nerve [axonal or myelin]), neuromuscular junction or muscle involvement.
- 2.
Temporal location of the origin of the problem: prenatal problems (genetically determined or disruptive), perinatal or postnatal (stroke, infection, etc.). Some problems do not have a specific temporal location: some cases of certain inherited metabolic diseases (IMD) and neurocutaneous syndromes combine central nervous system abnormalities that already began in the uterus.
- 3.
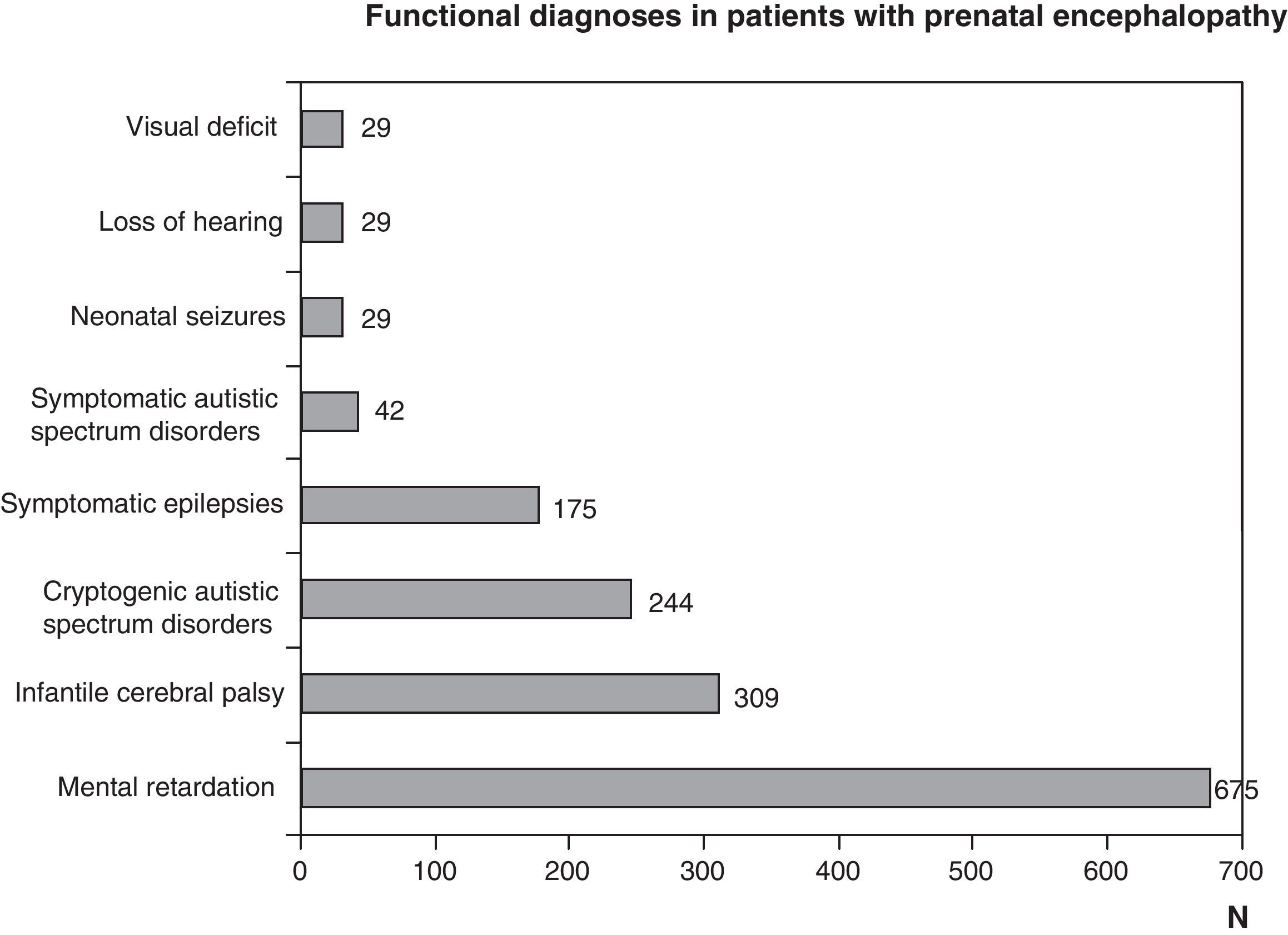
Functional diagnosis: motor problems through malfunctions at any level of the central nervous system, cognitive and behavioural problems, and epilepsy due to brain dysfunctions and sensory problems (vision or hearing) from encephalopathy or sight or hearing organ disorders. The most frequent functional diagnoses are childhood cerebral palsy, mental retardation, autism and epilepsy. They are all the result of encephalopathies, but could be due to different aetiologies, which could be prenatal, perinatal or postnatal. A pure motor problem could arise from an encephalopathy as well as a spinal cord or neuromuscular unit disorder. Encephalopathy may be seen as isolated disturbances or associated to motor, cognitive, behavioural or sensory (visual or hearing deficit) ones. Visual impairment and hearing loss may be caused solely by disturbances in the sensory organs, which in turn may be associated with encephalopathy or disturbances to other levels of the nervous system
- 4.
Aetiological diagnosis. This is what allows the diagnostic process to be closed, assesses therapeutic possibilities and establishes a prognosis and a risk of repetition in the family. Advances in the therapeutic and prenatal diagnosis and prevention areas are accompanied by a growing demand for early diagnosis.
It is frequently impossible to identify all these diagnostic levels. Moreover, the problems show different levels of severity; they can be isolated or several of them can be associated; they are evolutionary, and repercussions can be permanent or transitory.
A significant number of children monitored in neuropaediatrics present prenatal encephalopathies, and understood as those produced before the birth of the child. They can be disruptive (due to vascular problems during pregnancy, drugs or toxic substances or by congenital infections) and genetically determined, including IMDs.
From this perspective, we review the experience in the aetiological diagnosis of prenatal encephalopathies at our neuropaediatrics unit over the last 19 years.
Materials and methodsWe analysed the listed cases of prenatal encephalopathy, independent of their seriousness, which came from a neuropaediatric database from its inception on 15th May 1990 to 6th October 2005.1–5
The term encephalopathy is used in obedience to its etymological meaning of brain ailment, independently of whether it is extended or local and of its clinical repercussions. The diagnosis of prenatal encephalopathy has been established considering clinical and/or neuroimaging criteria. A prenatal origin of encephalopathy is supported by data such as the existence of polyhydramnios, dysmorphic facial features and associated extra-neurological malformations, as well as absence of perinatal or postnatal noxa. Prenatal encephalopathies are also considered to be cases of mental deficiency and cryptogenic autism. The identification by neuroimaging of agenesis of the corpus callosum, neuronal migration disorders or other prenatal abnormalities diagnoses prenatal encephalopathy. Cases with no evidence of prenatal encephalopathy such as regressive disorders, neurocutaneous syndromes and neuromuscular unit diseases were excluded.
We use the term cryptogenic according to its etymological meaning, applying it to problems of unknown cause.
Cases with a totally established aetiological diagnosis were collected together with those that did not need genetic or biochemical studies, such as cases of encephalopathy associated to myelomeningocele (hydrocephalus, Chiari anomaly type II and dysgenesis of the corpus callosum and others) or neurocutaneous syndromes with evidence of prenatal encephalopathy.
Also analysed were cases of prenatal encephalopathy with an unknown cause, their functional diagnoses, with a topographical location and their type of structural abnormality if applicable, as well as some of the studies carried out on them during their diagnostic process. We omitted some tests because they were not entered into the database until recently: skeletal series, abdominal ultrasound, cardiology study, oligosaccharides and glycosaminoglycans in urine.
ResultsDuring the study period of 19 years and 5 months, 11,910 children were included in the neuropaediatric database. Prenatal encephalopathy was diagnosed in 1596 (13.5%) cases. There were 1307 children (81.4%) who did not have a precise aetiological diagnosis. We have considered that from the total database there were 1480 children who had a rare disease, and are aware that many who do not have an established diagnosis also have rare diseases.
Table 1 presents the aetiological diagnosis established in 289 children where this has been possible.
Diagnoses established in prenatal encephalopathies.
| Chromosomal abnormalities (n=107) |
| Down's syndrome (n=33) |
| Fragile X syndrome (n=15) |
| Angelman syndrome (n=33) |
| Prader-Willi disease (n=7) |
| Subtelomeric deletion (n=7) |
| Edwards syndrome (n=7) |
| Patau syndrome (n=1) |
| Williams-Beuren syndrome (n=1) |
| Other chromosomal abnormalities (n=28) |
| Malformations in the central nervous system (n=59) |
| (Myelomeningocele with brain damage (n=31) |
| Genetic microcephaly (n=15) |
| Agyria/LIS1 gene mutation lissencephaly (n=3) |
| Joubert syndrome (n=2) |
| Walker-Warburg syndrome (n=1) |
| Inherited metabolic disease (n=34) |
| Mitochondriala disease (n=16) |
| Congenital deficiency of glycosylation (n=3) |
| Non-ketotic hyperglycinemia (n=3) |
| Cockayne syndrome (n=2) |
| GM1 gangliosidosis (n=1) |
| Hurler disease (n=1) |
| Mitochondrial beta-oxidation defect (n=1) |
| Merosin-deficient congenital muscular dystrophy (n=1) |
| Pelizaeus-Merzbacher disease (n=1) |
| Peroxisomal disorder (Zellweger variant) (n=1) |
| Smith-Lemli-Opitz (n=1) |
| Maple syrup disease (n=1) |
| 2-Methyl-3-hydroxybutyryl CoA dehydrogenase aciduria (n=1) |
| Biotinidase deficiency (n=1) |
| Disruptive encephalopathy (n=63) |
| Congenital infections (n=32) |
| Teratogenic encephalopathy (n=21): 16 FAS |
| Vascular: 9 twin (n=10) |
| Dysmorphic syndrome with mental retardation (n=20) |
| Neurocutaneous syndrome (n=10) |
| Congenital myotonic dystrophy (n=2) |
| Duchenne muscular dystrophy with mental retardation (n=1) |
FAS: foetal alcohol syndrome.
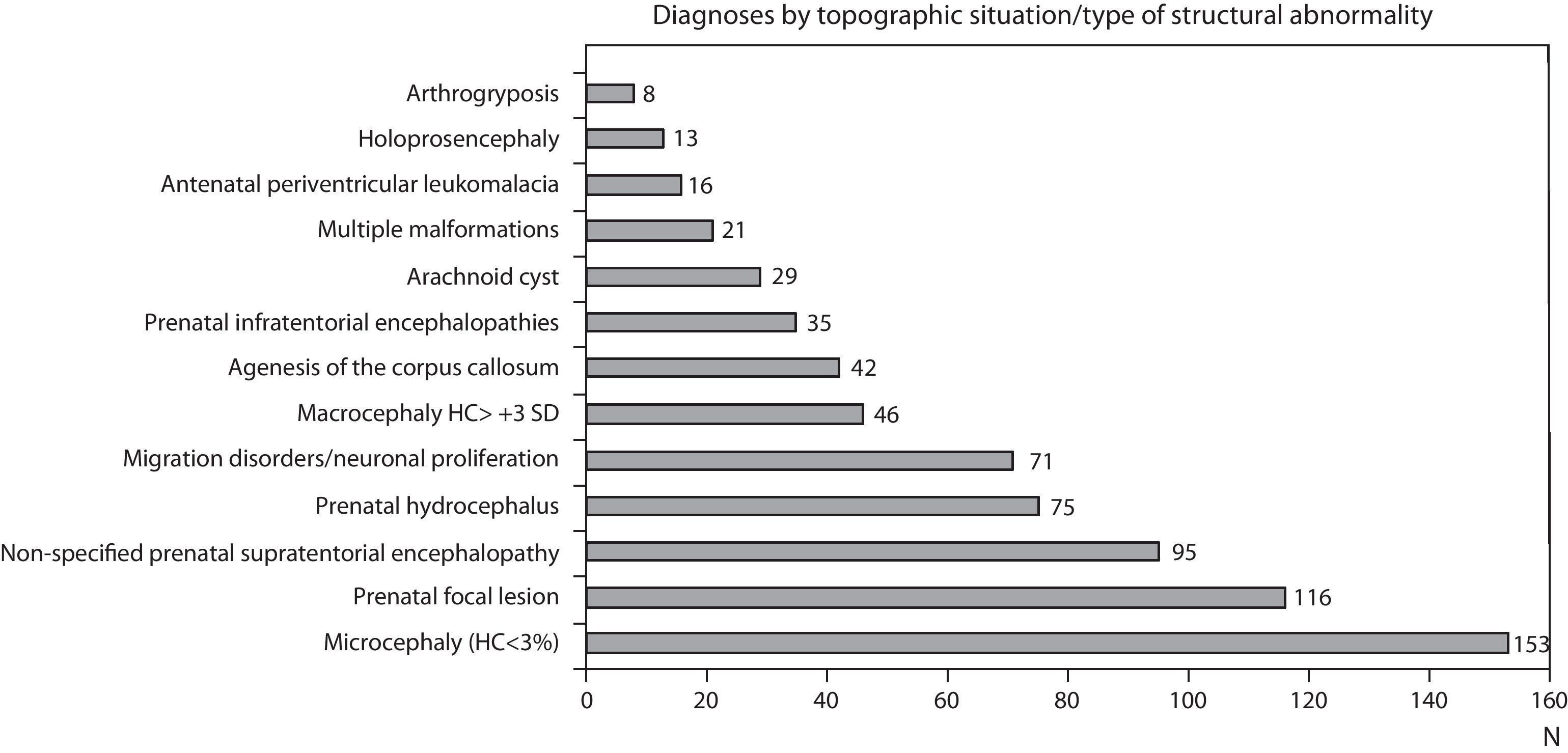
Figs. 1 and 2 show the most significant diagnoses for functional, topographic location and type of structural abnormalities assigned to the 1307 children with prenatal encephalopathy without an established aetiological diagnosis.


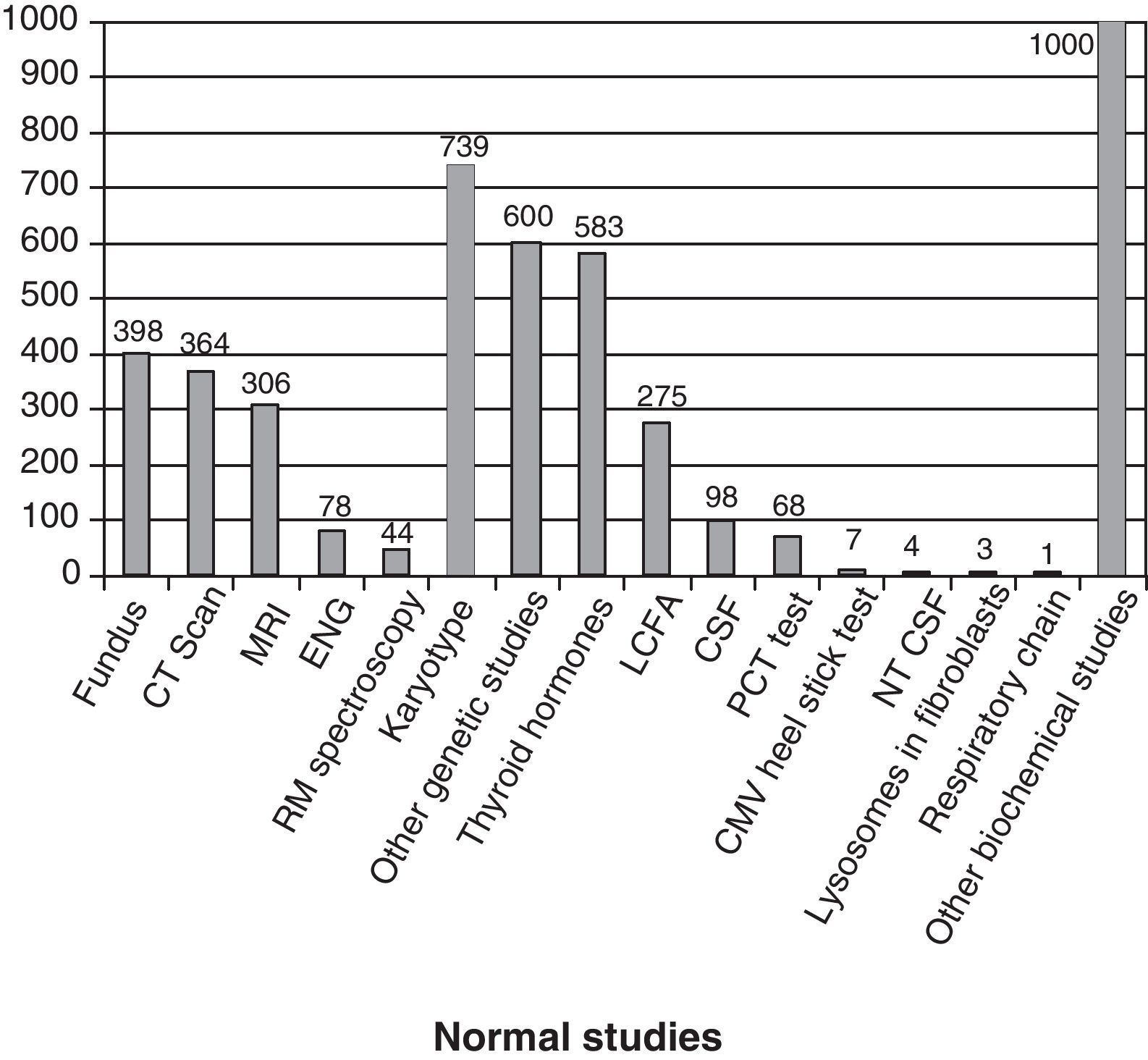
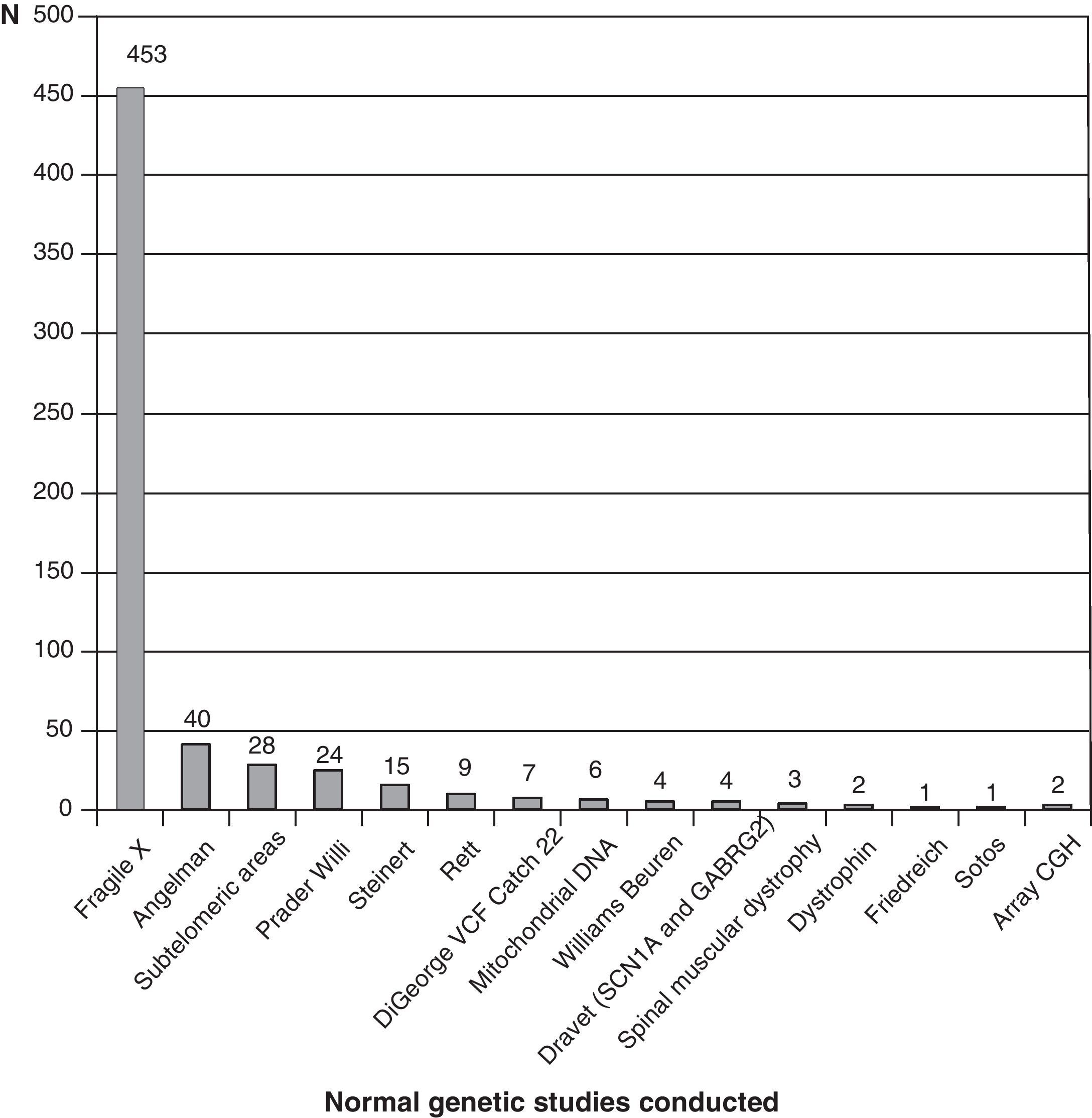
Fig. 3 shows the complementary studies carried out that have shown to be normal in the 1307 children without established aetiological diagnosis. Fig. 4 shows the normal genetic studies, different to karyotype, carried out. There were abnormal results that did not allow us to establish the aetiological diagnosis at least once in 320 children with a cranial CT scan, in 251 children with a brain MRI, in 35 children with fundus and in 6 children with electroneurogram.
. It this is altered, then the serum sialotransferrin pattern is determined.")
Normal studies carried out on the 1307 children without an established aetiology. We show the number of children with at least 1 normal study undertaken. LCFA: long-chain fatty acids; NTCSF: neurotransmitters in the cerebrospinal fluid; PCT test: percentage of poorly carbonated transferrin, which is useful in the identification of congenital disorders of N-glycosylation (CDG). It this is altered, then the serum sialotransferrin pattern is determined.

We admit that the defined concept of prenatal encephalopathy is vast and that there may be other disorders in it that involve dysfunctional brain areas, such as literacy or attention deficit disorders and hyperactivity; in these, the damage is less and it is generally accepted that complementary studies are not very cost-effective. In this study, we tried to analyse our real data compared to studies with established aetiological diagnoses in a wide and little homogeneous group with problems. The studies have therefore been carried out non-homogeneously in all patients, following clinical criteria and assets that have evolved over the period analysed, and that remain in constant adaptation.
Socio-sanitary demands increase with generalised access to information, active patient and family participation in controlling the disease, the increase of diagnostic possibilities and prenatal diagnosis and with increasingly good options for present or future treatment. The therapeutic possibilities of some lysosomal, mitochondrial and peroxisomal disorders (X-linked adrenoleukodystrophy, for example) enormously increase the case for early diagnosis.
The National Health System Strategy for rare diseases6 has recently been approved, totally in tune with the European Council Recommendations in the field of rare diseases.7 Second line strategy is prevention and early detection.
Many of the children included in this study (Table 1 and Figs. 1 and 2) are affected by rare diseases, whether or not they have been identified. Currently, rare diseases benefit from rapid advances in knowledge about them and they all have common general characteristics. They are very infrequent, even grouped among themselves; they are mainly hereditary, which means the need for genetic advice and study of families that could be potentially affected, as well as prenatal diagnostic strategies; they have a very complex diagnosis; many are very serious, chronic and disabling; and there is a great complexity of therapeutic options. Due to all of this, there is a great demand on the professionals involved and a need for multi-disciplinary teams to handle them.
We must point out that a high percentage of children with prenatal encephalopathy do not have an aetiological diagnosis (81%), despite the elevated number of studies undertaken (Figs. 3 and 4). However, these studies are necessary to identify many of the cases with established aetiological diagnosis. It is part of our work to monitor children without a diagnosis and it is not easy to find a balance between undertaking a lot of studies for few diagnoses or making do with the absence of a diagnosis.
Table 1 reflects that, in 10 out of 16 diagnoses of mitochondrial disease, this diagnosis is not assured, as there are partial deficits in the respiratory chain, in cases of unspecific clinical symptoms and with no other diagnostic supports. This lack of certainty made us carry out fewer studies after the increase in the mitochondrial disease diagnosis experienced a few years ago. In 2009, we proposed making a greater effort to diagnose genetic diseases and IMD.
Prenatal encephalopathies, genetically determined and disruptive, including many IMDs with a prenatal onset, can be clinically indistinguishable. Peroxisomal disorders, lysosomal and mitochondrial defects of glycosylation among other IMDs, congenital infections and chromosomal and gene abnormalities need specific studies for identification.8,10
Given the non-specificity of the signs and symptoms of many prenatal encephalopathies and prenatal or early onset IMD, an early diagnosis requires strategies for step-wise systematic studies, which must be permanently updated.9
Neuroimaging is useful in the study of prenatal encephalopathies; brain MRI is the most cost-effective one.11,12 Despite the need for anaesthesia, it has overtaken cranial CT scans due to the absence of ionizing radiation,13 a circumstance that is all the more important the younger the patient is.
In the systematics of prenatal encephalopathy study and prenatal and early onset IMD, we included the assessment of possible associated changes on an eye, cardiac, abdominal, nephro-urological, skeletal, muscular and peripheral nervous system level. The electroneurogram is especially useful during the first few years of life, where there may be a lack of clinical evidence of neuropathy, and in processes that associate first neuron disorders such as some leukodystrophies where the pyramidal clinical involvement could prevail over the neuropathy.9
Diagnostic possibilities increase with the availability of studies, especially biochemical and genetic ones.14,15 As well as those shown in Figs. 3 and 4, studies on congenital infection should also be carried out in some cases during the neonatal period, together with determinations of oligosaccharides and glycosaminoglycans and tests of guanidinoacetate and creatine in urine.
Genetic studies carried out on patients with no diagnosis (presumably genetic in many cases) are probably missing from this review, which could be due in part to the low cost-effectiveness of conventional karyotype. Genetic studies are constantly advancing and at a speed to which it is difficult to adapt. We have barely started to perform subtelomeric deletions when array CGH studies appear. The availability and cost-effectiveness of high resolution karyotype and array CGH tests are changing diagnostic orientation. Perhaps in the very near future, we will start with genetic studies and only if these are found to be normal will we consider other complementary tests.
Some IMDs have biological markers that orient the diagnosis. The serum sialotransferrin pattern and the PCT (percentage of poorly carbonated transferrin) test identify the majority of congenital defects in glycosylation. A pattern of long-chain fatty acids is found in the majority of peroxisomal disorders. Other biochemical data allow us to identify many IMDs, such as Menkes disease, Smith-Lemli-Opitz syndrome and Lesch-Nyhan syndrome, among others. However, there are cases where, after carrying out serial and even repeated tests, it is still not possible to determine if it is an IMD; we can then justify carrying out an ordered study, according to the greatest clinical suspicion, of the mitochondrial respiratory chain in muscle and skin and of certain lysosomal enzyme activities and pyruvate dehydrogenase deficiencies in cultured fibroblasts.
The strategies should abide by the principle of justice and should be applied in all cases in which they are specified.16,17 In many cases, these strategies do not exclude the individualised approach of each patient.
The diagnosis in some cases associates therapeutic possibilities; it nearly always allows the risk of repetition to be established, often associates prenatal and pre-implant diagnostic options and always gives answers to families and to us doctors as well, including in some cases the prognosis and approach of measures to prolong life, which are more difficult to establish with an absence of an established aetiological diagnosis.
It is difficult to establish limits and we do not have “evidence” that justifies these studies. We believe that the potential advantages of early diagnosis, including the savings in more tests, and prevention probably overcome the financial cost.9 Obviously, we should prioritise diseases that have therapeutic possibilities for diagnostic strategies.8,9,14
In any case, we need to make an effort to adapt to the growing demands of early diagnosis. Regrettably, the adaptability of neuropaediatric and IMD professionals as a group is becoming more and more limited: there are few of us and we have a heavy workload of healthcare and adaptation to growing demands. The public employment offer, which awards seniority, is in full confrontation with better public services, which should look for the best professionals with the ideal profile. These professionals need specific training, to be well paid and ideally to work exclusively in their full-time post. We have few possibilities of growth and improvement due to the lack of recognition in paediatric specialities and due to the health authorities’ approach and planning.
Conflict of interestThe authors have no conflict of interest to declare.
Presented at the 7th National Congress of Inborn Error of Metabolism, Bilbao, 22nd–23rd October 2009.
Please cite this article as: López Pisón J, et al. Encefalopatías prenatales. Nuestra experiencia diagnóstica de 19 años. ¿Hasta dónde con los estudios bioquímicos y genéticos? Neurología. 2011;26:481–7.
recomendados

Neurología (English Edition) sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas