






Primary central nervous system lymphoma is a rare subtype of extranodal non-Hodgkin lymphoma that accounts for 4% of central nervous system tumours.
Patients and methodsRetrospective review of 24 patients diagnosed with primary central nervous system lymphoma between 1990 and 2010. All patients were diagnosed using magnetic resonance imaging and the diagnosis was confirmed surgically.
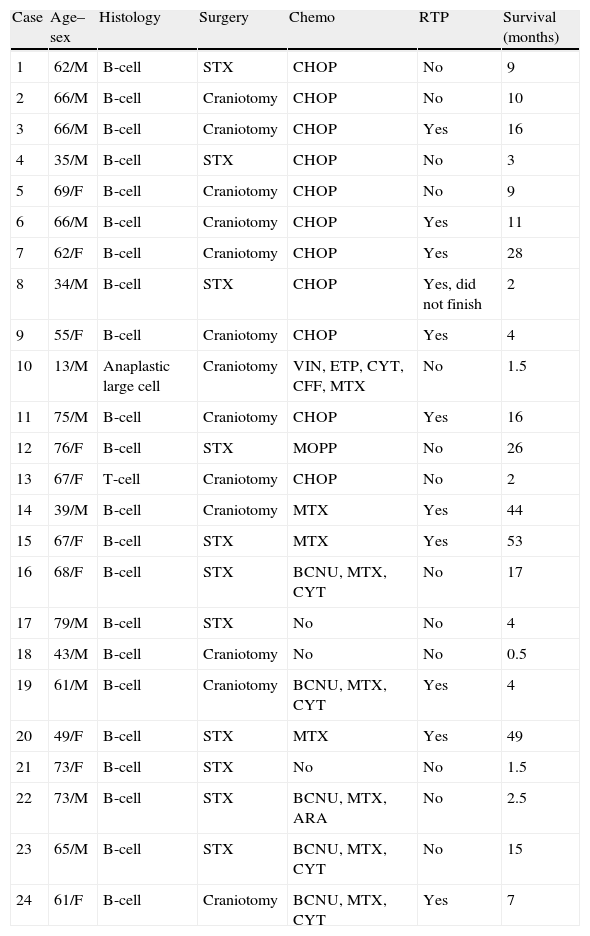
ResultsOf the 24 patients analysed, all except 4 were immunocompetent. Median age at diagnosis was 59.3 years (range 13–79) and the sex ratio (male to female) was 1:1.1. Cognitive decline (in 33.4%) and headache (in 25%) were the most common complaints. Diagnosis was performed in 13 cases (54%) following craniotomy and in the other 11 cases (46%) after stereotactic biopsy. Breakdown by pathology was as follows: 22 cases of B-cell lymphoma (91.6%), 1 case of anaplastic large-cell lymphoma, and 1 case of T-cell lymphoma. Mean survival time was 12.8 months with an overall 1-year survival rate of 37.5%.
ConclusionsPrimary central nervous system lymphoma often presents in the sixth decade with cognitive decline, headache, and focal neurological deficits. A single intracranial lesion was present in 75% of the patients (18 cases), and the remaining 25% (6 cases) had between 2 and 4 lesions. Preoperative clinical status was the most important factor determining prognosis.
Los linfomas primarios del sistema nervioso central son una variedad poco frecuente de linfomas no hodgkinianos que constituyen alrededor del 4% de los tumores del sistema nervioso central.
Pacientes y métodosrealizamos una revisión retrospectiva de 24 pacientes diagnosticados de linfoma primario del sistema nervioso central entre enero de 1990 y diciembre de 2010. Todos los pacientes fueron diagnosticados con resonancia magnética y confirmados quirúrgicamente.
ResultadosDe los 24 pacientes analizados, 4 presentaban inmunodeficiencia. La media de edad era de 59,3 años (intervalo 13-79) y la relación entre varones y mujeres de 1 a 1,1. El deterioro cognitivo (33,4% de los pacientes) y la cefalea (22,5%) fueron los signos de presentación más frecuentes. El diagnóstico se realizó en 13 casos (54%) tras llevar a cabo una craneotomía y en los otros 11 (46%) mediante biopsia estereotáctica. La distribución histológica mostró que 22 casos (91,6%) eran linfomas tipo B, un caso un linfoma anaplásico de células gigantes y el otro correspondió a un linfoma de células T. La supervivencia media fue de 12,8 meses y a un año del 37,5%.
ConclusionesLos linfomas cerebrales primarios se presentan alrededor de la sexta década de la vida y clínicamente se manifiestan con deterioro cognitivo, cefalea y déficits neurológicos focales. El 75% de los pacientes (18 casos) presentaban únicamente una lesión intracraneal y el restante 25% (6 pacientes) entre 2 y 4 lesiones. El estado clínico preoperatorio constituye el factor pronóstico más importante.
Primary central nervous system lymphoma (PCNSL) is a non-Hodgkin lymphoma originating in the cerebrum, eyes, leptomeninges, or spinal cord with no evidence of systemic lymphoma at the time of diagnosis. These tumours typically originate in B-cells and differentiating them from systemic non-Hodgkin lymphomas is difficult, whether by using a microscope or immunohistochemistry. In contrast, secondary brain lymphomas develop due to extension or spread of a systemic lymphoma in the central nervous system (CNS).1
PCNSLs account for about 4% of all primary brain tumours and between 1% and 2% of all lymphomas. The incidence rate of PCNSL has grown slowly in the past decades due to an increase in life expectancy in the general population and the presence of increasing numbers of immunocompromised patients.2
The first description,3 published by Bailey in 1929, referred to these tumours as ‘perivascular sarcomas’ since the cancer cells tended to surround blood vessels. In 1938, Yuile4 named them ‘reticular cell sarcomas’, while Russell and Rubinstein5 introduced the term ‘microgliomas’ in 1948. Their current name was coined by Henry et al.,6 who in 1974 differentiated primary CNS lymphomas from systemic lymphomas. Rappaport7 subsequently placed them in the group of non-Hodgkin lymphomas in his classification system.
We present a review of 24 patients diagnosed with PCNSL and treated in the last 21 years with a minimum follow-up time of 12 months. The study includes an analysis of patients’ clinical and neuroradiological characteristics and aspects related to treatment and progress.
Patients and methodsWe carried out a retrospective, descriptive study of patients diagnosed with primary brain lymphoma in our neurosurgery department between 1990 and 2010.
The study analyses patients’ demographic and clinical characteristics, neuroradiology diagnostic techniques used, surgical and oncological treatments employed, and patient progress. The Karnofsky Performance Scale (KPS) was used to assess patients clinically. In addition, we completed a systemic study using bone marrow analysis, abdominal ultrasound, and computed tomography (CT) to determine the tumour stage in all patients.
Tumours were confirmed in all cases using stereotaxic biopsy or craniotomy. All patients were monitored a minimum of 12 months.
ResultsThe series contains 24 patients (13 males and 11 females) with a mean age of 54.7 years (range, 13–79). In patients with AIDS, mean age was 37.7 years, while in all other patients, mean age was 60 (Table 1).
Histological characteristics, treatments administered, and survival of PCNSL patients.
| Case | Age–sex | Histology | Surgery | Chemo | RTP | Survival (months) |
| 1 | 62/M | B-cell | STX | CHOP | No | 9 |
| 2 | 66/M | B-cell | Craniotomy | CHOP | No | 10 |
| 3 | 66/M | B-cell | Craniotomy | CHOP | Yes | 16 |
| 4 | 35/M | B-cell | STX | CHOP | No | 3 |
| 5 | 69/F | B-cell | Craniotomy | CHOP | No | 9 |
| 6 | 66/M | B-cell | Craniotomy | CHOP | Yes | 11 |
| 7 | 62/F | B-cell | Craniotomy | CHOP | Yes | 28 |
| 8 | 34/M | B-cell | STX | CHOP | Yes, did not finish | 2 |
| 9 | 55/F | B-cell | Craniotomy | CHOP | Yes | 4 |
| 10 | 13/M | Anaplastic large cell | Craniotomy | VIN, ETP, CYT, CFF, MTX | No | 1.5 |
| 11 | 75/M | B-cell | Craniotomy | CHOP | Yes | 16 |
| 12 | 76/F | B-cell | STX | MOPP | No | 26 |
| 13 | 67/F | T-cell | Craniotomy | CHOP | No | 2 |
| 14 | 39/M | B-cell | Craniotomy | MTX | Yes | 44 |
| 15 | 67/F | B-cell | STX | MTX | Yes | 53 |
| 16 | 68/F | B-cell | STX | BCNU, MTX, CYT | No | 17 |
| 17 | 79/M | B-cell | STX | No | No | 4 |
| 18 | 43/M | B-cell | Craniotomy | No | No | 0.5 |
| 19 | 61/M | B-cell | Craniotomy | BCNU, MTX, CYT | Yes | 4 |
| 20 | 49/F | B-cell | STX | MTX | Yes | 49 |
| 21 | 73/F | B-cell | STX | No | No | 1.5 |
| 22 | 73/M | B-cell | STX | BCNU, MTX, ARA | No | 2.5 |
| 23 | 65/M | B-cell | STX | BCNU, MTX, CYT | No | 15 |
| 24 | 61/F | B-cell | Craniotomy | BCNU, MTX, CYT | Yes | 7 |
ARA: arabinoside; BCNU: carmustine; CFF: cyclophosphamide; CHOP: cyclophosphamide+doxorubicin+procarbazine+prednisone; CYT: cytarabine; ETP: etoposide; PCNSL: primary central nervous system lymphoma; MOPP: chlormethine+vincristine+procarbazine+prednisone; MTX: methotrexate; Chemo: chemotherapy; RTP: radiotherapy; STX: stereotaxic biopsy; VIN: vincristine.
The most common form of presentation was cognitive impairment in 8 patients (33.4%), followed by headache in 6 (25%), motor deficit in 5 (20.8%), and convulsions in the remaining 5 patients (20.8%). Four of the patients had AIDS.
According to the KPS, 14 patients scored between 90 and 100 at time of diagnosis, 6 scored between 80 and 70, and 4 patients scored between 60 and 50.
Diagnostic techniquesCT was used as the initial diagnostic method in all cases, and magnetic resonance imaging (MRI) was also used as a complementary method. CT results include 12 cases (50%) with hypodense areas, 8 (33.3%) with hyperdense areas, and 4 (16.7%) that were isodense. Contrast uptake was intense in 16 cases, including 3 with peripheral (ring-shaped) uptake; contrast enhancement was moderate in the remaining 8 cases.
In T1-weighted MRI sequences, tumours appeared hypointense in 13 cases (54%), isointense in 8 (33.3%), and moderately hyperintense in 3 (12.7%) with respect to grey matter. In T2-weighted sequences, 21 cases (87.5%) showed hyperintensities and the signal was heterogeneous in the remaining 3 (12.5%). Gadolinium uptake ranged from moderate to intense in all cases (Fig. 1).
Localisation Lesions adjacent to pia mater plane. (B) Single lesion. (C) Tumour in caudate nucleus. (D) Infiltration of the corpus callosum.")
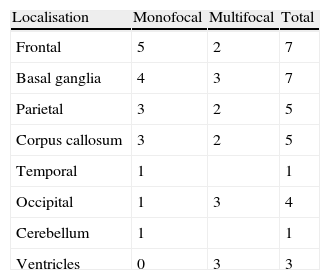
We identified a total of 33 intracranial lesions; 18 patients (75%) presented a single lesion, while 6 (25%) presented 2 to 4 lesions (Table 2). Right hemispheres were the most frequently affected, with 16 tumours (48.6%); 12 tumours (36.3%) were located in the left hemisphere, and the remaining 5 (15.1%) were in the corpus callosum (3 cases), cerebellar hemispheres (1 case), and lateral ventricles (1 lesion) (Fig. 2). The 4 patients with AIDS presented single tumours (2 lobar, 1 cerebellar, and 1 in the basal ganglia).
 CT with and without contrast showing a lesion in the right cerebellar hemisphere. (C and D) Contrast MRI corresponding to the same patient (Patient 18).")
Following histological diagnosis, our unit performed lumbar punctures on 18 patients to complete a biochemical and cytological study of CSF (the remaining 6 patients underwent oncological treatment in other hospitals). Abnormal spinal fluid protein levels were detected in 5 cases (range, 0.6–1.45g/L); 3 of these cases also displayed high cell counts (12–44 cells). Malignant cells were discovered in 5 out of 19 cases (26%).
TreatmentCraniotomy with wide-margin excision was performed in 13 patients (54%); total excision was achieved in 12 cases. Stereotaxic biopsy was used to establish the definitive diagnosis in the other 11 cases (46%) (Fig. 3).
Histology.")
Anatomical pathology studies found 22 B-cell lymphomas, 1 T-cell lymphoma, and 1 large-cell anaplastic lymphoma (Table 1).
Oncological treatmentDoctors provided oncological treatment to 21 patients (3 patients refused all types of complementary treatment). Courses of treatment varied widely (patients were treated in different regional haematology or oncology units), and treatments have also changed over the years. Patients included at the beginning of the study period followed the CHOP protocol (cyclophosphamide, doxorubicin, vincristine, and prednisone) or MOPP protocol (chlormethine, vincristine, procarbazine, and prednisone (1 case). The rest of the patients followed the different chemotherapy cycles listed in Table 1; all cycles included methotrexate (MTX).
Radiotherapy was used to treat 14 patients and ruled out for 5 patients due to their poor clinical status and low probability of survival (cases 4, 10, 13, 18, and 21). The remaining 5 patients refused radiotherapy. The dose ranged from 42 to 50.5Gy and radiation was limited to the cranial area in all cases.
Course of diseaseAt the time we reviewed these medical histories (November 2011), 23 patients had died; only 1 patient aged 49 years was still alive after a follow-up time of 49 months. Mean survival time was 12.8 months (range, 0.5–53). Among the 4 patients with AIDS, mean survival time was 12.2 months (range, 2–44); in the rest of the patients, it was 15.5 months (range, 0.3–64). Nine patients (37.5%) were still alive after 1 year of follow-up.
In patients younger than 55, mean survival was 16.6 months, vs 13 months in patients aged 55 and older. Regarding clinical stages, mean survival time in patients with a Karnofsky score ≥80 was 21.2 months, vs 7.1 months in the rest of the patients.
DiscussionPCNSLs account for approximately 4% of all primary brain tumours and represent 1% to 2% of all malignant non-Hodgkin lymphomas. However, up to 10% of cases of lymphoma at any location may present with neurological impairment of some type.8 The incidence rate of this type of tumour has risen slowly in recent decades to reach 30 cases/106inhabitants/year.1 This makes PCNSL the second most common malignant brain tumour in the United States after glioma.9 On the one hand, this increase in incidence is caused by the growing number of immunocompromised patients, including transplant recipients, cancer patients, and those with AIDS. Another major factor contributing to increased incidence is prolonged life expectancy, given that PCNSL cases have tripled among patients older than 60, and the explanation for this tendency has yet to be made clear.1,9,10 Researchers calculate that for AIDS patients, the risk of developing PCNSL at some point during the disease ranges from 2% to 6%. Among allograft recipients treated with immunosuppressant drugs, the risk ranges from 1% to 5%, and risk for patients suffering from congenital immune deficiencies is calculated to be 4%.11 A surprising finding from our study is that none of the patients had received immunosuppressant drugs. We cannot provide a clear explanation for this situation, although it is possible that patients in this category may have had poor chances of survival, which would have ruled out performing a brain biopsy.
No significant sex differences were found for incidence, although most series, like our own, report slight male predominance. This may be due to AIDS incidence being higher in men. Peak incidence is found in the sixth decade, with a smaller peak in the third to fourth decade that corresponds to patients with immune deficiencies.1
Presentation of PCNSL resembles that of other expansile intracranial processes, although cognitive decline and headache are more frequent in lymphomas. Other symptoms, such as neurological deficits or convulsions, may also appear. Between 10% and 15% of all patients may present with ocular symptoms (uveitis or intraocular lymphoma). In our series, only 1 young patient experienced vitreous infiltration at 18 months after onset.
In CT studies, PCNSLs appear as single or multiple tumours that are round or oval-shaped, well-defined, and typically hyperdense.12 They are surrounded by hypodense areas that correspond to oedema.13 Moderate to pronounced enhancement was observed after administration of contrast. Centrally located tumours are more likely to have homogeneous uptake, while peripherally located tumours show ring-shaped enhancement. This uptake pattern is the most common among infratentorial tumours.14 Although our 4 cases with AIDS had single lesions, immunocompromised patients were more likely to present lesions at multiple locations (up to 50% of these cases). Where multiple lesions were present, they tended to be more invasive.15 Meningeal infiltration is found in 75% of these cases,16 and such cases must not be confused with primary meningeal tumours or cranial lymphomas that progress to infiltrate the meninges.17
MRI shows meningeal infiltration as hypointense areas in T1-weighted sequences and isointense areas in T2-weighted sequences compared to grey matter, although other types of signals are not uncommon.18 Administering gadolinium provokes pronounced enhancement; the contrast marks the tumour margins and separates the solid nucleus from adjacent oedematous tissue with no contrast uptake.15
Preoperative diffusion imaging studies showed that diffusion restriction was present in 90% of patients. Although diffusion restriction may also be observed with gliomas, metastasis, and other expansile processes, its presence in PCNSL is very pronounced and the apparent diffusion coefficient value is lower.19 A recent study by Barajas et al.20 indicates that the apparent diffusion coefficient prior to treatment may have predictive value for this type of lymphoma. MR spectroscopy revealed a decrease in N-acetylaspartate concentration and elevated values for lipids, choline, and the choline/creatinine ratio.20,21 Positron emission tomography (PET) studies have a diagnostic sensitivity of 100% regardless of the method used.22 PET studies typically reveal hypermetabolic lesions in these cases.20 When hypermetabolism is observed in immunocompromised patients with suspected infectious or parasitic disease, it is a key factor in performing differential diagnosis.20
Macroscopically, hypermetabolic lesions are tumours resembling high-grade gliomas, and they also present with infiltrating and necrotic areas.1 Tumours may be located in either grey or white matter; in more than 80% of all cases, they are located in deep grey matter in the supratentorial area and tend to appear near the ventricular system. Microscopically, they are composed of masses of lymphoid cells with high cell density in the central parts of the tumour where the structure of the cerebral parenchyma disappears. Cells group around blood vessels, occupying and widening the Virchow-Robin spaces; this process separates reticular fibres and induces new fibre formation.23 Although these masses are well-defined, it is not uncommon to find neoplastic invasion beyond the macroscopic tumour margins. According to Isaacson and Norton,24 75% to 90% of these tumours are diffuse large B-cell lymphomas (centroblastic or most of all, immunoblastic; 92% in our series). Burkitt lymphomas account for 5% and another 10% to 25% are low-grade lymphomas, especially lymphoplasmacytoid lymphomas, with diffuse follicle centre lymphomas being less common.
Four pathogenic patterns have been described.
- I.
Solitary nodule (56%) or multiple nodules (26%) in intracranial locations. This is the most common form.6,15,25
- II.
Diffuse meningeal impairment or periventricular lesions that infiltrate the subarachnoid space in 20% to 50% of all cases.26 They may also cause ependymal invasion.27
- III.
Vitreous or uveal deposits (15%).28,29
- IV.
Intradural spinal tumours.30–32 This last type is extremely rare and accounts for less than 1% of all PCNSL cases.
Most immunophenotypes express typical pan-B-cell antigens (CD20 and CD79a) with monoclonal expression of surface immunoglobulins, especially IgMk. PCNSLs associated with immunosuppression tend to express latent membrane proteins or Epstein–Barr virus nuclear antigens, unlike what occurs in immunocompetent patients. T-cell lymphomas tested positive for CD45RO and CD3.8 Some studies show that BCL-6 overexpression is variable and tends to be associated with a better prognosis. In turn, p57 and C-Myc expression signal a poorer prognosis.33
In molecular genetic studies, lymphomas express clonal rearrangement of the immunoglobulin gene (for B-cell lymphomas) or the T-cell receptor gene (for T-cell lymphomas). Researchers have described losses of genetic material as occurring mainly at chromosome 6. These losses are related to poorer prognosis; the most common gains are located at 12q. Methylation of the promoter of the reduced folate carrier gene has also been linked to MTX resistance.34
Given that lymphoid tissue is not present in the nervous system, the pathogenesis of these lymphomas remains a topic for debate. The numerous hypotheses are all speculative given that few evidence-based studies are available.35 One explanation is that lymphoma cells can originate anywhere in the body (outside the CNS), but they may develop in the brain after receiving homing receptors specific to brain endothelium. Once they are established in this tissue, the immune syndrome is unable to destroy them. Another theory states that there must be a pre-existing inflammatory lesion that would elicit a polyclonal response from lymphoid cells. This in turn could produce a neoplastic clone, which also occurs with other types of lymphomas.11 A third hypothesis is that while lymphoma cells generated outside the CNS will be eradicated systematically by a functional immune system, they proliferate within the brain.15
Treatment begins after the surgical procedure has delivered a diagnosis. In our study, craniotomy was performed in 13 cases (54.1%) and stereotaxic biopsy in 11. In most series, however, diagnosis is performed using biopsy, since lesions are often found in deep and/or multiple locations. Furthermore, studies have been unable to confirm a better prognosis with wide-margin excision than with a biopsy. We must stress that the latter technique is less invasive and possesses a lower rate of complications than craniotomy.36,37 It also has a diagnostic accuracy of more than 95%. Tumour resection by craniotomy may give rise to neurological impairment and delays in beginning oncological treatment in addition to not improving survival.38,39 This technique is only indicated for easily accessible single lesions in which the procedure will not increase morbidity. When stereotaxic biopsy is performed, reducing steroid treatment is important as its cytolytic effect may decrease tumour size. On some occasions, tumours have even disappeared completely (phantom tumour).40 Although some authors state that establishing a good diagnosis without withdrawing steroid treatment is possible,41 general consensus is that steroids should be discontinued 5 to 10 days before the biopsy. In each of our cases, we took samples after at least 10 days of having discontinued steroids. We were able to establish a definitive diagnosis based on the first biopsy in all cases.
After diagnosing the tumour, doctors must determine lymphoma extension. Impairment of different areas of the CNS, including the eyes, meninges, and cranial nerves, does not imply either a more advanced stage or a poorer prognosis.38 It is calculated that between 4% and 12% of lymphomas initially classified as PCNSLs have systemic effects.42 While CSF cytology is listed by all protocols as a test for lymphoma extension, it was only positive in 27.7% of the patients in our series. Meningeal involvement increases the positivity rates of the results.43 While CSF analysis is important for diagnostic orientation, it also has significant implications for treatment. Intrathecal presence of tumour cells justifies the use of intrathecal treatment.8 An MRI scan of the entire neuroaxis must be performed if doctors have not done so previously. PET scan tracking may come to replace the procedure using thoracic-abdominal CT and bone marrow biopsy. Studies indicate that it is more sensitive for detecting small systemic lymphoma foci.22,44 Although spontaneous remission of PCNSL without prior steroid treatment has been described,45 these cases are exceptional, and we believe that such events do not constitute a reason to delay treatment. Oncological treatment is based on corticosteroid treatment, chemotherapy, and radiotherapy, as tumours are particularly sensitive to these types of treatment.1
Corticosteroids induce cytolysis in PCNSL cells and may cause partial or total response in 40% of immunocompetent patients. Nevertheless, these benefits are short-lived; the treatment does not resolve the illness or predict a better outcome. Combined treatment with chemotherapy and radiotherapy is currently believed to be the best approach. Traditional systemic lymphoma protocols (CHOP) are not effective because they do not pass the blood–brain barrier (BBB).38 Although treatment with CHOP elicits a good initial response since most of the tumour is not protected by the BBB, subsequent cycles of CHOP will not completely eradicate the remaining part of the lymphoma. This is probably due to the BBB reverting to a normal state after the initial doses of treatment.38 Our study reports use of widely differing treatment protocols. This is due, firstly, to the considerable length of the study period, and secondly, because 6 of the patients were referred to our unit by other hospitals in our region for purposes of undergoing craniotomy or biopsy. After the procedure, they were treated at their own local hospitals.
Cytarabine and MTX are the most active drugs for immunocompetent patients. Treatment schedules that include both of those drugs and radiotherapy have a response rate approaching 80% and mean survival of 3 years.14
Trials with temozolomide (TMZ) are currently underway. The advantages of this drug are good tolerance and a 1-year complete remission rate of 31% in patients who do not respond to MTX treatment.46 Combined therapy with high doses of TMZ and MTX is delivering promising results, even in elderly patients.47 Monotherapy with TMZ is associated with a 1-year complete remission rate of 47% and mean survival of 21 months in elderly patients.48
Rituximab is a very commonly used treatment for peripheral lymphomas, but as a macroprotein, it has poor BBB penetration. A few studies of rituximab monotherapy or combined rituximab-MTX treatment have been published.49 Although the level of evidence for using rituximab in PCNSL is low, some oncologists consider it beneficial. In general, however, doctors believe that it should only be used in prospective trials.38
Some authors recommend intra-arterial chemotherapy preceded by mannitol to elicit a transient disruption in the BBB and therefore increase the level of drugs in the CNS.50
Although there are no comparative studies, retrospective analysis of the series employing intrathecal MTX did not show better survival compared to series that did not use that treatment.51 In a recent retrospective study by Sierra et al.52 analysing 69 patients with PCNSL in which 39 patients were treated with MTX and the other 30 were not, there were no statistically significant differences between the 2 groups.
Neurotoxicity is a serious problem for patients on combined treatment with chemotherapy and radiotherapy. Most patients develop instability, loss of sphincter control, and cognitive decline; MRI scans show cerebral atrophy and diffuse impairment of the white matter. In patients older than 60, the risk of neurotoxicity is nearly 100% and radiotherapy is not recommended if PCNSL is in remission at the end of the course of chemotherapy. Recently, Correa et al.53 observed that cognitive impairment is more pronounced in surviving patients treated with high doses of MTX and radiotherapy than in those only receiving high doses of MTX. A recent study by Alimohamed et al.54 describes an alternative; in this study, 21 patients with PCNSL were treated with high-dose thiotepa, busulfan, cyclophosphamide and autologous hematopoietic stem cell transplantation without holocranial radiotherapy. These doctors observed that none of the patients developed neurotoxicity and that 52% remained alive and disease-free at 60 months.
Radiotherapy is one of the pillars of treating PCNSL. Full cranial radiotherapy is indicated since local radiation is associated with higher recurrence rates. Spinal radiation is not recommended even if CSF dissemination is present because it increases morbidity without improving survival.39 This treatment is only indicated in cases of spinal lymphoma, which are rare.1 Radiotherapy as sole treatment rarely produces remission; mean survival in cases in which this approach was used ranged from 10 to 18 months.55 However, it may be indicated as palliative treatment or in patients with small lymphocytic or lymphoblastic lymphomas originating in the meninges.38 Although radiotherapy doses vary from study to study, they generally range between 40 and 50Gy. Using doses higher than 50Gy with an additional superimpression has not been shown to be more effective, and this practice also increases toxicity.38
Another controversial subject is how to treat relapses. There is no definitive strategy at present and the treatment approach should consider the patient's age, functional status, and prior treatments. In younger patients with a good general status, rescue therapy including autologous transplant may be applied. A study by Soussain et al.56 describes 43 patients with relapsing PCNSL who were treated with 2 cycles of cytarabine and etoposide followed by autologous stem-cell transplant. These researchers observed a 2-year survival rate of 69% (76% for those who underwent transplant while in complete remission).
A recent, multi-centre study by Thiel et al.57 analysed 551 immunocompetent patients with PCNSL. Patients were randomly assigned to 2 groups; group 1 was treated with high-dose MTX and radiotherapy, while the other group was treated with high-dose MTX in monotherapy. They observed that patients treated with radiotherapy developed more toxicity; however, no significant intergroup differences were detected with regard to disease-free time or survival.
The prognosis for CNS lymphomas is much worse than for systemic lymphomas.58 Mean survival without treatment is 2 to 4 months following diagnosis.59 With radiotherapy, mean survival reaches 10 months; combined with chemotherapy and intrathecal MTX, initial response rates are as high as 85% and recurrences tend to appear after 15 to 45 months of treatment.1 Patients with AIDS in our series had a slightly shorter survival time than the other patients; this tendency is largely explained by the stage of progression of the underlying disease.1
In our experience, the patient's clinical status prior to surgery was the most important prognostic factor. Patients with a Karnofsky score ≥80 had survival times up to 3 times longer than those with scores below that threshold. Although the literature indicates that outcomes are poorer in patients with AIDS, our study finds little difference between the survival times in each of the groups (12.2 vs 15.3 months). These results should be interpreted with caution, however, as the group size is very small. The literature identifies 5 independent factors predicting prognosis: age, KPS, elevated lactate dehydrogenase (LDH) in serum, elevated protein in CSF, and involvement of deep regions.60 The International Extranodal Lymphoma Study Group distinguishes between 3 risk levels according to how many of the factors named above are present (0–1, 2–3, or 4–5).61 Although opinions differ between authors, factors indicative of good prognosis include single intracranial lesion, absence of meningeal or periventricular tumours, no immune deficiency, and treatment with both chemotherapy and radiotherapy.62,63
There are 2 incidence peaks for PCNSL in the fourth and sixth decades of life; the former corresponds to patients with AIDS. The disease's most frequent form of clinical presentation is cognitive decline, followed by headaches and focal neurological impairment. In our study, 75% of the patients presented single intracranial lesions. Most PCNSLs fell into the histological category of large B-cell lymphomas and clinical status prior to surgery was the most important prognostic factor.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Gelabert-González M, et al. Linfomas primarios del sistema nervioso central. Neurología. 2013;28:283–93.
