



Progressive multifocal leukoencephalopathy (PML) is a demyelinating disease of the central nervous system caused by reactivation of a latent infection with JC virus. We report the case of a patient diagnosed with systemic lupus erythematosus (SLE) and receiving low-dose prednisone.
Our patient was an 83-year-old man with hypertension and no other relevant medical history, who since the age of 81 years had been under follow-up by the rheumatology department due to symptoms of arthritis and probable SLE (antinuclear antibodies, lymphopaenia, thrombocytopaenia). Prednisone at a maximum dose of 10 mg/day achieved satisfactory control of the articular symptoms, but lymphopaenia persisted.
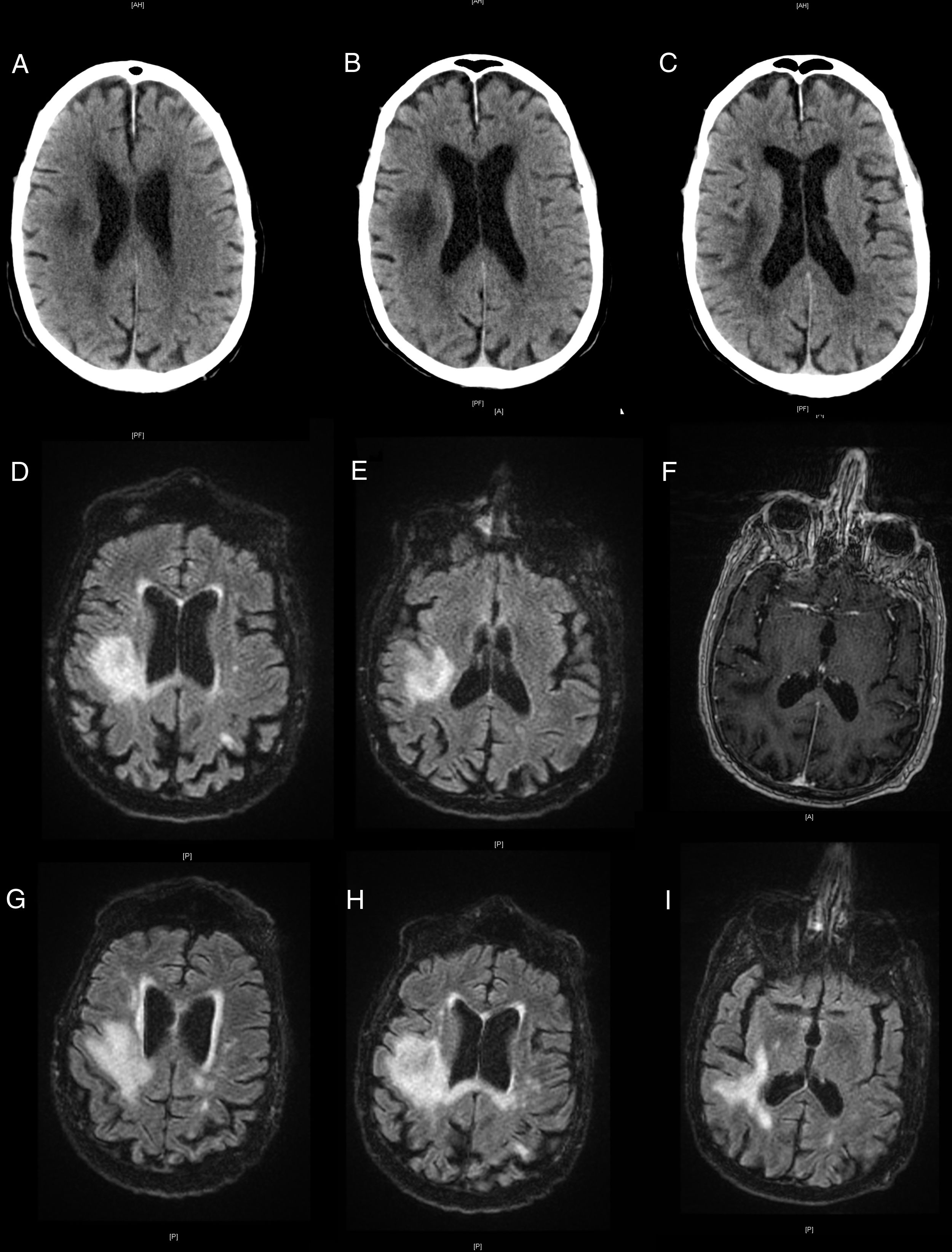
The patient visited the emergency department due to left hemiparesis. A head CT scan revealed a hypodense lesion in the right frontoparietal area (Fig. 1A), of probable ischaemic origin; antiplatelet treatment was started. He returned a week later due to symptom progression; the examination revealed visual extinction in the left visual hemifield and left hemiplegia and hemihypaesthesia. A further head CT scan (Fig. 1B and C) revealed extension of the previously detected lesion.
, and progression a week later (B and C). Brain MRI scan showing a hyperintense lesion on T2-weighted FLAIR sequences (D and E) and no contrast uptake on gadolinium-enhanced T1-weighted sequences (F). Progression of the lesions on T2-weighted FLAIR sequences (G-I).")
Neuroimages showing the progression of the lesion. Non-contrast head CT scans showing a hypodensity in the right hemisphere (A), and progression a week later (B and C). Brain MRI scan showing a hyperintense lesion on T2-weighted FLAIR sequences (D and E) and no contrast uptake on gadolinium-enhanced T1-weighted sequences (F). Progression of the lesions on T2-weighted FLAIR sequences (G-I).
The patient was admitted for further testing. A brain MRI scan revealed a lesion in the right frontoparietal region, which was hypointense on T1-weighted sequences and hyperintense on T2-weighted sequences, with no contrast uptake (Fig. 1D-F). An emergency biochemical and cytological analysis of a CSF sample showed no pathological findings. We suspected inflammatory aetiology due to the chronology of the symptoms and our patient’s history, and started empirical treatment with methylprednisolone at 1 g/day while we awaited laboratory results.
Our patient worsened, presenting bulbar symptoms and a low level of consciousness. An additional brain MRI scan revealed lesion growth, with meningeal involvement and contrast uptake in right cranial nerves IX and XI (Fig. 1G-I). Laboratory analyses revealed lymphopaenia, with 237 CD4 T cells/mm3 and a high CD4/CD8 ratio. CSF findings were negative for oligoclonal bands and positive for JC virus, which led to diagnosis of PML. Active treatment was ruled out given the patient’s age and clinical status, and he died 6 weeks after symptom onset.
The seroprevalence of JC virus is 65% among individuals older than 17 years, and even higher among those above the age of 70.1 Virus reactivation in the form of PML has become more frequent in recent decades due to increased prevalence of HIV and the more widespread use of immunosuppressants. However, virus reactivation has also been described in patients with minimal immunosuppression2 and even in immunocompetent individuals.3 The risk of developing PML is greater in patients with SLE than in those with other rheumatic disorders.4,5 Up to 40% of cases present in patients with minimal iatrogenic immunosuppression2,6 and no other risk factors for PML.5
This gives rise to the hypothesis that SLE may increase the risk of developing PML, although the mechanisms are not fully understood. The striking increase in the incidence of PML during the HIV/AIDS epidemic and the presence of PML in patients with idiopathic CD4 lymphopaenia underscores the significant role of CD4 T cells in controlling JC virus infection.7 In both scenarios, CD4 lymphopaenia is detected in the peripheral blood; CSF lymphocyte levels in these patients are not well documented, as in the case presented here.
It has recently been suggested that 2 forms of lymphopaenia may present in patients with SLE: the more frequent form is associated with the activity of SLE, and may improve with immunosuppressants, whereas the other form is more sustained, is not related to disease activity, and is associated with greater risk of infection, since it behaves as an immunodeficiency disorder.8 Given the shortage of information on the immune status of patients with PML and their treatment history,6 it may be interesting to analyse whether sustained CD4 lymphopaenia or its association with a specific drug9 increases the risk of the infection. This may enable the development of management algorithms,8 like those used with multiple sclerosis.10
In any case, PML is rare in patients with SLE, with an estimated prevalence of 4 cases per 100 000 patients (0.44% of all cases of PML),5 although it is probably underdiagnosed. The condition should be suspected in patients with SLE who present progressive neurological impairment and white matter lesions with no mass effect or contrast uptake.11 This diagnosis may improve prognosis since it enables early discontinuation of immunosuppressant treatment; this is currently the most effective management strategy.11
Our case highlights the risk of PML in patients with SLE and minimal iatrogenic immunosuppression who present sustained CD4 lymphopaenia. The study of treatment and immunophenotypic profiles of patients with SLE and PML may enable the development of more individualised treatment algorithms aiming to revert lymphopaenia, decreasing the incidence of this devastating entity.
These authors contributed equally to the manuscript.
Please cite this article as: Casado L, Hervás C, Quintas S, Vivancos J. Leucoencefalopatía multifocal progresiva en paciente con lupus eritematoso sistémico: ¿podría ser la linfocitopenia-CD4+ el principal factor de riesgo? Neurología. 2020;35:667–669.
recomendados

Neurología (English Edition) sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas