




El síndrome de Moyamoya es un trastorno idiopático caracterizado por la oclusión progresiva de las arterias carótidas internas intracraneales asociada con la formación bilateral de una red de pequeños vasos (fenómeno de Moyamoya), que se sitúan alrededor del área estenótica1. Entre las entidades que se asocian a este síndrome están la anemia de células falciformes, la neurofibromatosis tipo 1, la radioterapia cerebral y el síndrome de Down2. La neurofibromatosis tipo 1 (NF1) es un trastorno autosómico dominante caracterizado por la aparición de manchas café con leche, efélides axilares, neurofibromas cutáneos, nódulos de Lisch, displasias óseas y gliomas ópticos. Cuando el síndrome de Moyamoya se asocia a la NF1 las manifestaciones cerebrovasculares suelen aparecer en la edad infantil3.
Mujer de 47 años, hipertensa, fumadora y migrañosa. Sin antecedentes familiares de enfermedades cerebrovasculares ni de trastornos neurocutáneos. Antecedentes cerebrovasculares: 3 años antes presentó un cuadro de hemiparesia derecha y afasia leve secundaria a ictus isquémicos a nivel de ambos ganglios basales y frontoparietal izquierdo de etiología no determinada. Se realizó estudio analítico extenso, ecocardiograma y Doppler de troncos supra-aórticos que no mostraron alteraciones significativas. En la arteriografía cerebral se observó una oclusión de la arteria cerebral media derecha desde su segmento M1 con circulación colateral a través de ramas meníngeas de la cerebral anterior y territorio profundo. En el segmento M1 de la arteria cerebral media izquierda se apreciaba una estenosis de algo menos de 1cm, así como en 2 zonas del segmento M2 de la división superior que presentaban características arrosariadas. No existían datos evidentes de red de pequeños vasos a nivel del área estenótica. Se pautó tratamiento con aspirina® a dosis de 300mg al día y prednisona en pauta descendente quedándose con una dosis de 10mg al día. Al alta persistía una hemiparesia derecha leve y leve afasia nominal.
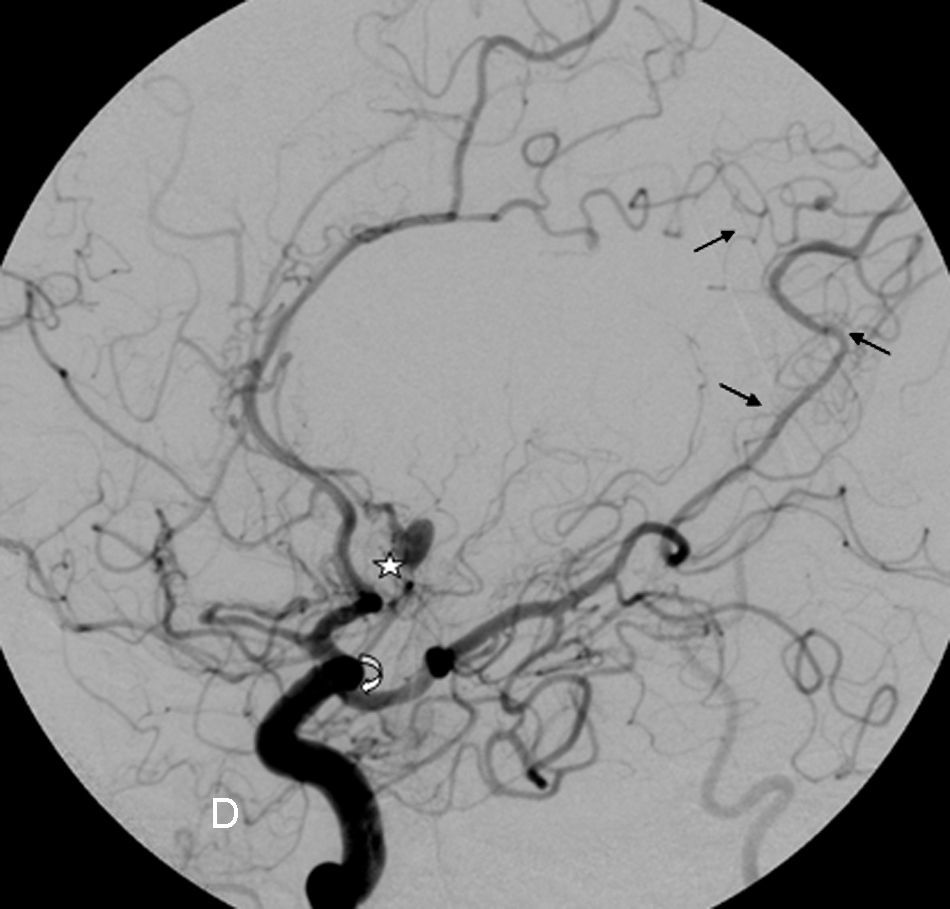
Ingresa por cefalea súbita y disminución del nivel de conciencia objetivándose en el TAC craneal una hemorragia subaracnoidea más evidente a nivel de zona medial de ambos lóbulos frontales. La exploración física exhaustiva evidencia varias lesiones café con leche a nivel abdominal y muslo izquierdo, efélides axilares y fibromas cutáneos que cumplían con los criterios diagnósticos de NF1 (tabla 1). La paciente presentó complicaciones de crisis comiciales e hidrocefalia obstructiva, pero se desestimó la colocación de una válvula de derivación. Se realizó arteriografía cerebral (fig. 1) objetivándose oclusiones arteriales de ambas arterias cerebrales medias con recanalizaciones leptomeníngeas a través de durosinangiosis, compatible con enfermedad de Moyamoya, así como la existencia de seudoaneurisma del segmento A2 de la arteria cerebral anterior izquierda. La paciente presentó un brusco empeoramiento a los 15 días del ingreso con resangrado subaracnoideo que le condujo a la muerte. No se realizó estudio necrópsico por negativa familiar.
Criterios diagnóstico de la neurofibromatosis tipo 1
| Para el diagnóstico de neurofibromatosis tipo 1 el paciente debe tener al menos 2 de los siguientes 7 criterios: |
| 1. Seis o más manchas café con leche iguales o mayores de 5mm en pacientes prepúberes y de 15mm de diámetro en pacientes pospúberes |
| 2. Dos o más neurofibromas de cualquier tipo, o uno plexiforme |
| 3. Presencia de pecas en axilas o ingles |
| 4. Glioma del nervio óptico |
| 5. Dos o más nódulos de Lisch (hamartomas del iris) |
| 6. Lesión ósea definida como displasia del esfenoides o adelgazamiento de la cortical de los huesos largos con o sin seudoartrosis |
| 7. Un familiar de primer grado afecto (padres, hermanos o hijos) según los criterios previos |
Fuente: Tomado de la National Institute of Health Consensus Development Conference6.
. El territorio silviano se encuentra revascularizado a través de la cerebral anterior y posterior, así como de la carótida externa por la arteria meníngea media (durosinangiosis: flechas rectas) e imagen de aneurisma del segmento A2 de la arteria cerebral anterior izquierda (estrella).")
Arteriografía cerebral. Imagen del territorio carotídeo derecho. Oclusión completa de la arteria cerebral media derecha en su origen en la bifurcación carotídea, con una estenosis de la carótida supraclinoidea (flecha curva). El territorio silviano se encuentra revascularizado a través de la cerebral anterior y posterior, así como de la carótida externa por la arteria meníngea media (durosinangiosis: flechas rectas) e imagen de aneurisma del segmento A2 de la arteria cerebral anterior izquierda (estrella).
El diagnóstico del síndrome de Moyamoya se basa en los hallazgos angiográficos: estenosis u oclusión uni o bilateral de la arteria carótida interna intracraneal o las ramas proximales del polígono de Willis en fases precoces; múltiples colaterales pequeñas lenticuloestriadas y talamoperforantes con aspecto de bocanada de humo o nube de humo (Moyamoya en japonés) en la fase intermedia; y anastomosis trasdurales o transóseas entre la carótida interna y externa en fases tardías4.
En una serie extensa de 70 pacientes con infartos cerebrales de etiología inhabitual, solamente un único paciente presentó un síndrome de Moyamoya, lo que incide en lo infrecuente de esta etiología y más aún su asociación a NF15. Nuestra paciente cumple con los criterios diagnósticos de NF16. Puede faltar la existencia de antecedentes familiares en los casos de mutaciones de novo como en nuestra paciente. Estudios de ligamiento han encontrado que uno de los genes de la enfermedad de Moyamoya familiar se localiza en el cromosoma 17q25, que está en estrecha relación con el gen de la NF1 situado en 17q11.27. Esto sugiere la posibilidad de alelismo de estos 2 genes8,9. Los pacientes con hallazgos unilaterales se consideran que presentan un síndrome de Moyamoya, incluso si no se asocian a otros trastornos8. Estudios longitudinales evidencian una afectación contralateral en hasta un 40% de los pacientes que inicialmente se presentan con afectación unilateral10. La historia natural de este trastorno es variable. La progresión de la enfermedad puede ser lenta, con escasos episodios intercurrentes o fulminante con un rápido declive neurológico11. Un estudio efectuado en el año 2005 indicó que el porcentaje de progresión es alto incluso entre los pacientes asintomáticos, y que el tratamiento médico (antiagregantes, estatinas, antihipertensivos y abandono del hábito tabáquico) no detiene la progresión de la enfermedad12.
Mientras que los ictus isquémicos se desarrollan en niños, la hemorragia subaracnoidea suele aparecer en pacientes adultos13. La hemorragia intracraneal típicamente afecta a los adultos con este trastorno. La localización de la hemorragia puede ser intraventricular, intraparenquimatosa (generalmente a nivel de los ganglios basales) o subaracnoidea. El sangrado subaracnoideo se ha atribuido a la rotura de las colaterales frágiles que aparecen conforme progresa la estenosis de la carótida interna. Cambios en los patrones circulatorios en la base del cerebro se han implicado en la aparición de aneurismas cerebrales que también contribuyen a la mayor incidencia de hemorragia subaracnoidea que presentan estos pacientes13. Estudios patológicos han puesto de manifiesto que las arterias afectas no muestran cambios ateroscleróticos ni vasculíticos que produzcan la oclusión14, sino que esta se produce por la combinación de hiperplasia de células musculares lisas y trombosis intraluminal15.
En conclusión, en pacientes jóvenes con ictus isquémicos bilaterales siempre hay que pensar en la enfermedad de Moyamoya que puede confirmarse realizando mediante una arteriografía donde se evidencia la estenosis de arterias carótidas intracraneales junto al desarrollo de múltiples colaterales pequeñas alrededor del área estenótica. Del reconocimiento precoz de esta entidad y la realización del tratamiento quirúrgico de revascularización depende el pronóstico. Por último, en pacientes con un síndrome de Moyamoya se debe llevar a cabo una exploración cutánea exhaustiva en busca de lesiones dermatológicas típicas de la NF1.

