



El síndrome obesidad de rápida progresión, disfunción hipotalámica, hipoventilación alveolar y disregulación autonómica (ROHHAD) es una entidad infrecuente y compleja con comienzo en niños sanos a los 2-4 años, donde además un 40% se relaciona con tumores de la cresta neural.
DesarrolloPresentamos el caso de una niña que comenzó a los 2 años con un cuadro de obesidad de rápida progresión y posteriormente asoció disfunción hipotalámica con trastornos electrolíticos graves, trastorno de conducta, hipoventilación y disautonomía graves, entre otros. Aunque su fisiopatología no está aclarada, una de las hipótesis actuales del ROHHAD es autoinmune y, tras respuesta limitada a inmunoglobulinas por vía intravenosa, se decidió probar respuesta a ciclofosfamida a dosis altas (a dosis bajas tampoco fue eficaz). Esto motivó múltiples complicaciones graves posteriores que requirieron estancia prolongada en la UCI (a destacar mielinólisis central pontina recuperada e imposibilidad de destete del respirador, requiriendo traqueotomía para continuar asistencia respiratoria). Aunque la conducta mejoró, el desenlace fue fatal a los 5 años debido a un episodio de muerte súbita en domicilio por su patología respiratoria.
ConclusionesSe trata de una patología poco conocida que precisa un abordaje multidisciplinar, dada la complejidad de los síntomas y su implicación multisistémica. Es necesario identificar precozmente la hipoventilación alveolar y el inicio de tratamiento adecuado por su implicación pronóstica. La experiencia con inmunomoduladores como inmunoglobulinas, ciclofosfamida o rituximab muestra una mejoría de los síntomas en algunos casos. Sería deseable realizar estudios multicéntricos, dada la baja incidencia del síndrome, para esclarecer su fisiopatología y diseñar su adecuado abordaje terapéutico.
ROHHAD syndrome (rapid-onset obesity with hypothalamic dysregulation, hypoventilation, and autonomic dysregulation) is a rare and complex disease, presenting in previously healthy children at the age of 2-4 years. Up to 40% of cases are associated with neural crest tumours.
DevelopmentWe present the case of a 2-year-old girl with symptoms of rapidly progressing obesity, who a few months later developed hypothalamic dysfunction with severe electrolyte imbalance, behaviour disorder, hypoventilation, and severe autonomic dysregulation, among other symptoms. Although the pathophysiology of this syndrome remains unclear, an autoimmune hypothesis has been proposed for ROHHAD. Therefore, after obtaining a limited response to intravenous immunoglobulins, we decided to test the response to a high dose cyclophosphamide (low dose was not effective either). Unfortunately our patient experienced many severe complications (among them central pontine myelinolysis, from which the patient recovered, and failure to wean from the ventilator requiring tracheostomy and long term ventilation) that required a prolonged ICU stay. Although her behaviour improved, our patient unfortunately died suddenly at home at the age of 5 due to respiratory pathology.
ConclusionsROHHAD syndrome is a rare and little-known disease which requires a multidisciplinary approach because it involves complex symptoms and multiple organ system involvement. Alveolar hypoventilation should be identified early and appropriate treatment should be started promptly for the best possible outcome. Immunomodulatory treatment with immunoglobulins, cyclophosphamide, or rituximab has previously resulted in symptom improvement in some cases. Because of the low incidence of the syndrome, multi-centre studies must be carried out in order to gather more accurate information about ROHHAD pathophysiology and design an appropriate therapeutic approach.
El trastorno conocido en la literatura anglosajona como rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation (ROHHAD) es un trastorno muy poco frecuente que incluye anomalías del sistema endocrino (principalmente del hipotálamo), del sistema nervioso autonómico y respiratorio (hipoventilación central). Hasta la fecha, hay descritos menos de 100 pacientes en todo el mundo. Aunque existe referencia de una paciente española de 1996 que podría cumplir los criterios actuales1, se presenta el primer caso español confirmado con síndrome ROHHAD.
El ROHHAD se ha solapado durante años y puede haberse confundido en la literatura más antigua con el síndrome de hipoventilación central congénito de inicio tardío (later-onset congenital central hypoventilation syndrome [LO-CCHS]). La descripción de mutaciones en el gen PHOX2B como criterio diagnóstico2,3 y la ausencia de estas en los pacientes con ROHHAD4-6 permitieron establecer la distinción entre estas 2 entidades nosológicas.
El término ROHHAD, así como los criterios diagnósticos, fueron inicialmente descritos por Ize-Ludlow et al.4. El primer síntoma suele ser hiperfagia, en niños previamente normales hasta los 2-4 años, que provoca un aumento exagerado de peso en poco tiempo (10-15kg en 6-12 meses). Progresivamente, en los meses y años siguientes van apareciendo otras alteraciones de la función hipotalámica, estando entre las más frecuentes la alteración del metabolismo hidrosalino, junto con síntomas de disautonomía.
Además, los pacientes presentan apnea obstructiva del sueño de forma precoz y posteriormente hipoventilación alveolar central, siendo el manejo de esta última uno de los principales factores pronóstico. Los trastornos de conducta y del desarrollo cognitivo y del lenguaje son muy frecuentes. Aproximadamente el 40% de los casos descritos presentan además a lo largo de su evolución tumores de la cresta neural (ganglioneuromas y ganglioneuroblastomas), que pueden aparecer incluso varios años después del inicio de la sintomatología4-6. De ahí que la tendencia actual sea a llamar a esta entidad ROHHAD-NET (neuro-ectodermal tumors).
Se presentan nuestra experiencia con el primer caso de síndrome ROHHAD publicado en español y la revisión de la literatura disponible.
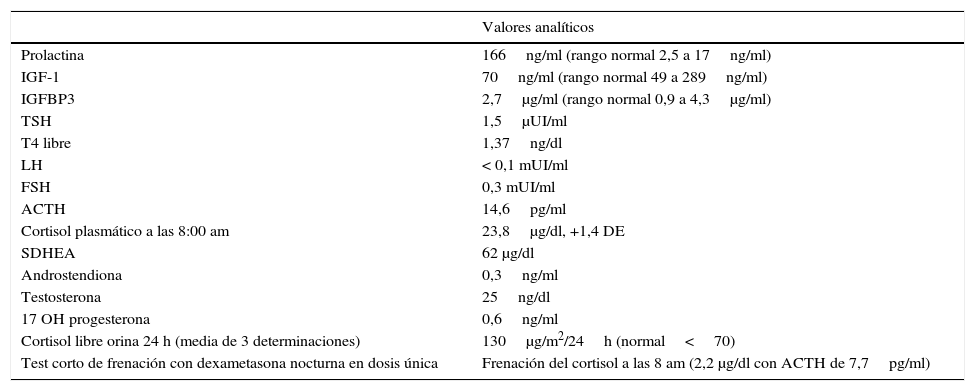
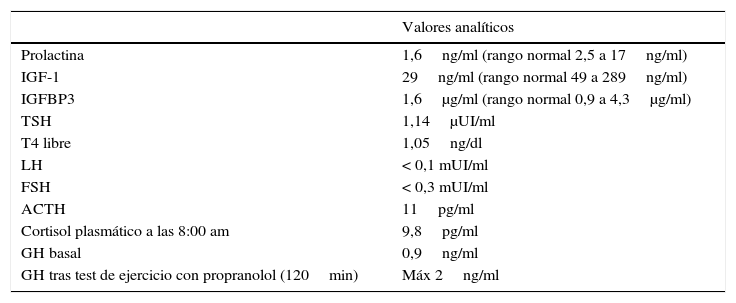
Caso clínicoNiña remitida desde Atención Primaria a los 2 años y 10 meses por un cuadro de obesidad de rápida progresión (8kg en 10 meses) junto con un gran aumento del apetito. El embarazo, el parto y el período neonatal fueron normales. El desarrollo pondero-estatural y psicomotor también habían sido normales. El peso se encontraba en+3,8 DE, la talla en –0,8 DE con una velocidad de crecimiento normal y acorde con su talla diana (–0,5 DE) y el IMC era de+6,8 DE. La exploración era normal sin estigmas cushingoides ni acantosis nigricans y la presión arterial era normal. Era la primera hija de unos padres jóvenes y sanos no consanguíneos. Dentro de los estudios de laboratorio realizados, la bioquímica sanguínea y urinaria fue normal. En el estudio hormonal basal destacaba únicamente hiperprolactinemia grave y persistente (166ng/ml, rango normal 2,5-17ng/ml), que orientaba al diagnóstico de adenoma hipofisario o de patología hipotalámica. Los resultados de las analíticas hormonales a esta edad se resumen en la tabla 1. Se descartó hipercortisolismo primario y dentro del estudio de hipercortisolismo de origen central, dada la hipercortisoluria y la hiperprolactinemia de repetición, se solicitó una resonancia magnética nuclear (RM) cerebral e hipotálamo-hipofisaria con contraste, que no detectó ninguna alteración. Dos meses después apareció estrabismo convergente bilateral, realizándose estudio de fondo de ojo, pares craneales y con exploración neurológica normales. Poco después presentó empeoramiento importante de la conducta con episodios de auto y heteroagresividad, hiperactividad e impulsividad, que fueron refractarios a tratamiento con neurolépticos (risperidona, aripiprazol y olanzapina). Llamaba la atención la baja sensibilidad al dolor, ya que la niña se golpeaba la cabeza con fuerza sin quejarse e incluso sonreía durante las venopunciones. Se le realizó una RM de tórax y abdomen sin observar ningún hallazgo patológico, así como el estudio genético de síndrome de Prader-Willi y Smith-Magenis, que fueron negativos. A los 3 años y 3 meses presentó el primer episodio de alteración del metabolismo hidrosalino con hipernatremia grave (175 mEq/l), sin hallazgos indicativos de diabetes insípida. Posteriormente, precisó múltiples ingresos tanto por hiper como hiponatremia, algunos coincidiendo con infecciones y otros sin aparente desencadenante. Se indicó a los padres un control estricto de la ingesta de líquidos, sin presentar nunca claros datos analíticos de déficit ni de exceso de producción de hormona antidiurética (ADH). Durante uno de los ingresos se constataron episodios de apnea/hipopnea, principalmente durante el sueño, y los padres referían episodios de cianosis en domicilio. Además, comenzó a presentar signos de disregulación autonómica: episodios de hipotermia (hasta 32°C) sin desencadenante aparente, tendencia a la bradicardia y alteraciones de la motilidad digestiva (reflujo gastroesofágico grave desde los 3 años, que precisó funduplicatura de Nissen y prolapso rectal completo de primer grado, que requirió rectopexia). El peso, que inicialmente había aumentado muy rápidamente, se normalizó alrededor de los 3 años debido a las dificultades en la nutrición. En este momento, dadas la evolución y la presencia de los síntomas y signos descritos, se llegó a la sospecha diagnóstica de síndrome ROHHAD. Se le solicitaron autoanticuerpos en suero y en líquido cefalorrarídeo (LCR), que incluían: anti-NMDA, anti-AMPA y anti-GABA-B, que fueron negativos. También se le realizó estudio de errores congénitos del metabolismo (aminoácidos, ácidos orgánicos, Saicar, sulfitest, lactato/piruvato y neurotransmisores en el LCR) con resultado normal. El cariotipo y el estudio molecular del gen PHOXB2 también fueron normales. En el momento del diagnóstico, se decidió iniciar tratamiento con inmunoglobulina por vía intravenosa (IV) a una dosis de 2g/kg con frecuencia mensual, el cual se mantuvo durante 8 meses, durante los cuales no se apreció mejoría de la conducta, pero tampoco presentó ningún episodio de hiper o hiponatremia graves, ni tampoco de hipotermia. Endocrinológicamente, a partir de los 4 años se detectó un enlentecimiento de la velocidad de crecimiento (talla –2 DE, peso –0,2 DE, IMC+1,2 DE y VC de –3,5 DE) con diagnóstico de déficit de hormona de crecimiento. Los estudios analíticos realizados se resumen en la tabla 2. A los 5 años se detectó hipotiroidismo central (TSH 0,19, T4L 0,2ng/dl), iniciándose tratamiento sustitutivo con tiroxina a 3 μg/kg/día, presentando normalización secundaria de los niveles de IGF-I e IGF-BP3 (52ng/ml y 1.500μg/ml, respectivamente). Posteriormente, asoció insuficiencia suprarrenal crónica de origen central (cortisol basal 1,3 μg/dl con respuesta límite en test de ACTH, 18 μg/dl a los 60 min de la administración de 250 μg de ACTH sintética), iniciándose tratamiento con hidrocortisona por vía oral a 13mg/m2/día. Tras este se observó una mejoría de la bradicardia, la somnolencia, la hipotermia y la tendencia a la hipoglucemia leve, aunque persistía el trastorno de conducta. Ante la aparición de nueva sintomatología, y dada la sospecha de participación autoinmunitaria en la fisiopatología de la enfermedad, se decidió iniciar tratamiento con ciclofosfamida a dosis de 750mg/m2/dosis mensual durante 4 meses, apreciando mejoría leve y transitoria tras cada ciclo. Por este motivo, siguiendo la experiencia de Paz-Priel et al.7 con una paciente publicada y 2 más no publicados, en los que se había asociado mejoría de la conducta con una pauta de ciclofosfamida a dosis altas (50mg/kg/día durante 4 días consecutivos), se decidió iniciar este tratamiento teniendo la niña en ese momento 5 años de edad. Coincidiendo con la neutropenia posterior a ciclofosfamida, presentó un empeoramiento brusco y desarrolló un cuadro de sepsis de origen respiratorio por Pseudomonas aeruginosa, que precisó ingreso en cuidados intensivos pediátricos, prolongándose durante casi 4 meses por múltiples complicaciones en su recuperación. A la inestabilidad hemodinámica que presentaba la paciente por shock séptico, se sumaron síntomas de disautonomía ya apreciados con anterioridad, como son bradicardia e hipotermia, complicando el cuadro de shock actual. Se decidió inicio de tratamiento con corticoides ante shock refractario a catecolaminas (hidrocortisona 100mg/m2). Tras lenta recuperación de la aplasia medular, la paciente superó el cuadro agudo, aunque siguió siendo dependiente de ventilación mecánica tras múltiples intentos de extubación (atelectasias masivas y persistencia de apneas de origen central). Asimismo, se constata un cuadro de tetraparesia espástica con afectación de pares craneales bajos con deterioro del nivel de consciencia, por lo que se realizó una RM cerebral, en la que se objetivaron signos de mielinólisis central pontina. Es cierto que durante su ingreso había presentado numerosos episodios de alteración del metabolismo hidrosalino de muy difícil manejo, atribuibles a la disfunción hipotalámica. Así, alternaba períodos de diabetes insípida (DI), en especial en la fase de shock, con otros con síndrome de secreción inadecuada de hormona antidiurética (SIADH), siendo estos últimos más frecuentes. En los períodos de DI llegó a alcanzar valores de sodio de 175 mEq/l máximo, que precisaron control estricto de balance y desmopresina. En los períodos de predominio de SIADH se hizo restricción de líquidos, junto con uso de diuréticos, alcanzando cifra mínima de sodio de 123mg/dl. Una vez superado el proceso agudo, la impresión general de los médicos a su cargo fue que el manejo «más fisiológico» era el más estable para la paciente, evitando en la medida de lo posible fluidoterapia por vía IV, en especial con fluidos hipotónicos, e intentos de manejo con aportes de nutrición y líquidos vía enteral para evitar hiponatremia. Aunque no se objetivaron cambios bruscos de natremia u osmolaridad, probablemente su alteración hipotalámica de base pudo predisponer el desarrollo de la mielinólisis central pontina durante el proceso agudo. Tras la revisión de la literatura disponible8,9 se decide prueba de tratamiento con inmunoglobulinas por vía IV (0,4g/kg dosis durante 5 días), con buena evolución en las semanas siguientes, recuperando progresivamente el nivel de consciencia, iniciando articulación de lenguaje, movilidad espontánea de las 4 extremidades, siendo capaz de masticar y deglutir e incluso deambular. En este momento se decidió realizar traqueotomía para continuar su asistencia respiratoria y finalmente se logró el alta a domicilio con seguimiento por la unidad de hospitalización domiciliaria y múltiples consultas de especialidad. La disautonomía evolucionó, llegando a precisar implantación de marcapasos por bradicardia sinusal extrema a los 6 meses del alta y además es llamativo que, a pesar de presentar procesos infecciosos, no solía presentar fiebre, ya que tendía a la hipotermia (basal de 35°C).
Valores hormonales al diagnóstico (edad 2 años+10 meses)
| Valores analíticos | |
|---|---|
| Prolactina | 166ng/ml (rango normal 2,5 a 17ng/ml) |
| IGF-1 | 70ng/ml (rango normal 49 a 289ng/ml) |
| IGFBP3 | 2,7μg/ml (rango normal 0,9 a 4,3μg/ml) |
| TSH | 1,5μUI/ml |
| T4 libre | 1,37ng/dl |
| LH | < 0,1 mUI/ml |
| FSH | 0,3 mUI/ml |
| ACTH | 14,6pg/ml |
| Cortisol plasmático a las 8:00 am | 23,8μg/dl, +1,4 DE |
| SDHEA | 62 μg/dl |
| Androstendiona | 0,3ng/ml |
| Testosterona | 25ng/dl |
| 17 OH progesterona | 0,6ng/ml |
| Cortisol libre orina 24 h (media de 3 determinaciones) | 130μg/m2/24h (normal<70) |
| Test corto de frenación con dexametasona nocturna en dosis única | Frenación del cortisol a las 8 am (2,2 μg/dl con ACTH de 7,7pg/ml) |
Valores hormonales al diagnóstico (edad 4 años)
| Valores analíticos | |
|---|---|
| Prolactina | 1,6ng/ml (rango normal 2,5 a 17ng/ml) |
| IGF-1 | 29ng/ml (rango normal 49 a 289ng/ml) |
| IGFBP3 | 1,6μg/ml (rango normal 0,9 a 4,3μg/ml) |
| TSH | 1,14μUI/ml |
| T4 libre | 1,05ng/dl |
| LH | < 0,1 mUI/ml |
| FSH | < 0,3 mUI/ml |
| ACTH | 11pg/ml |
| Cortisol plasmático a las 8:00 am | 9,8pg/ml |
| GH basal | 0,9ng/ml |
| GH tras test de ejercicio con propranolol (120min) | Máx 2ng/ml |
Aproximadamente a los 9 meses de la administración del tratamiento con ciclofosfamida la niña acudía al colegio y realizaba sus tareas (antes era completamente dependiente de sus cuidadores), controlaba esfínteres, comía sola, deambulaba de modo autónomo, jugaba con otros niños, recuperó la sensibilidad para el dolor y era capaz de mantener una conversación sencilla coherente. Aunque persistía cierto déficit cognitivo, así como secuelas motoras (signos piramidales leves), su conducta mejoró considerablemente (únicamente necesitaba risperidona a dosis bajas), así como su calidad de vida y la de su familia. Entre otras, la niña continuaba seguimiento por Oncología para el despistaje de tumores y desde el punto de vista respiratorio continuaba siendo dependiente de oxigenoterapia de forma intermitente y precisaba ventilación mecánica a través de traqueotomía por apneas centrales e hipoventilación alveolar durante el sueño. La afectación respiratoria es la principal determinante del pronóstico del síndrome de ROHHAD y por desgracia, a los 6 años, la niña presentó un episodio de muerte súbita en domicilio, sin que en ese momento se encontrase conectada a respirador y tras constatar los servicios de emergencias buena función del marcapasos. Los padres no concedieron autopsia.
DiscusiónEl síndrome ROHHAD es un cuadro extraordinariamente complejo que requiere para su diagnóstico de un alto grado de sospecha y precisa de manejo necesariamente multidisciplinar, que incluya la colaboración de especialistas en Endocrinología, Psiquiatría, Cirugía, Neumología, Oncología, Neuropediatría, Cardiología y otros. Además, aunque el tratamiento sea conservador, ha de iniciarse de forma precoz para mejorar su pronóstico e intentar evitar sus complicaciones. Haciendo un breve repaso a la historia de este síndrome, el primer caso de ROHHAD fue descrito en 1965 por Fishman et al.10, asociando a su fenotipo característico hipoventilación central de origen tardío y disfunción hipotalámica. No es hasta el año 2000 cuando Katz et al.11 establecen una clara diferencia con el CCHS, haciendo una descripción en detalle de un caso y revisando otros 10 pacientes publicados previamente. En su artículo exponen una lista de signos y síntomas comunes, que se sigue usando hasta la fecha. En 2007 Ize-Ludlow4 y su grupo confirman la ausencia de la mutación del gen PHOX2B en los casos con ROHHAD y definen su nombre, aunque la nueva nomenclatura ROHHAD-NET introducida en los últimos años incluye su fuerte asociación a este tipo de tumores6. Nosotros hemos preferido no usar dicha terminología por estar mucho menos extendida y hemos preferido la clásica nomenclatura de ROHHAD. Su diagnóstico es clínico y por exclusión, puesto que actualmente la etiología continúa sin estar aclarada12. Probablemente, el síndrome esté asociado a cierta predisposición genética junto con la participación de factores paraneoplásicos o inmunológicos, pero no se ha podido localizar ninguna alteración genética específica, no se han identificado cambios epigenéticos ni hay ninguna evidencia de su asociación con ningún fenómeno autoinmune. En 1995 se introduce la idea de una posible asociación paraneoplásica o autoinmune13, pero esta teoría ha de tomarse con prudencia, puesto que hasta la fecha solo está descrita la experiencia de expertos con tratamiento inmunomodulador o inmunosupresor en casos individuales o series de casos7,14,15. Tras hacer una revisión de la literatura disponible, cabe mencionar que se ha descrito la presencia de infiltración linfocitaria e histiocitaria en el hipotálamo de algunos pacientes13 y en los últimos años se ha publicado su posible asociación con la presencia de biomarcadores como bandas oligoclonales en el LCR16. También se han planteado recientemente dudas sobre su posible implicación genética, o al menos monogenética, tras presentarse en solo una niña de una pareja de gemelas monocigóticas12. Aunque podría tratarse de genes modificadores, este último hallazgo hace más fuerte la posible asociación con otras etiologías. Para su diagnóstico se ha sugerido que entre los criterios clínicos debe constar ganancia rápida de peso e hipoventilación de aparición a partir del año y medio de edad. Además, los pacientes deben asociar disfunción hipotalámica con al menos uno de los siguientes: obesidad de rápida evolución, hiperprolactinemia, hipotiroidismo central, alteraciones del metabolismo hidrosalino, fracaso del test con estimulación con hormona de crecimiento, déficit de corticotropina o alteración del inicio de la pubertad.
Para asegurar el diagnóstico, se deberá excluir además CCHS y síndrome de Prader-Willi15. Como se ha descrito previamente, los niños con ROHHAD tienen un desarrollo y crecimiento normales hasta el inicio de los síntomas entre los 1,5 y 9 de edad, progresando en los años siguientes. El primer síntoma suele ser la ganancia de peso, asociando posteriormente disfunción hipotalámica4, como en el caso que describimos. Es llamativa la evolución endocrinológica de nuestra paciente, que presentó inicialmente un patrón indicativo de seudo-Cushing e hiperprolactinemia, evolucionando a hipofunción hipotálamo-hipofisaria generalizada (hipotiroidismo e insuficiencia suprarrenal centrales, disminución de la IGF I, prolactina y FSH/LH). Además, asoció oscilaciones hormonales previamente descritas en este síndrome, como la alteración del metabolismo hidrosalino con episodios tanto de hiper como hiponatremia, sin datos claros de disfunción en la secreción de ADH. En nuestro caso, y a diferencia de la literatura previa, el peso evolucionó satisfactoriamente, teniendo en su última revisión un IMC en +0,14 DE, en probable relación con la magnitud del reflujo gastroesofágico, las numerosas complicaciones a lo largo de su evolución y los ingresos prolongados. En cuanto a la talla, es posible que el retraso del crecimiento (talla –3,38 DE) se hubiera normalizado progresivamente tras la instauración del tratamiento del hipotiroidismo central. La afectación respiratoria asociada al síndrome ROHHAD ocurre tarde o temprano en su evolución1,4,15,17. Inicialmente, los pacientes presentan apnea obstructiva del sueño y con los años todos presentan hipoventilación alveolar central. Esta última se caracteriza por la ausencia de signos de dificultad respiratoria asociada a hipercapnia y/o hipoxemia, dando lugar a progresiva ventilación inadecuada, que sumada a la disautonomía coexistente supone un alto riesgo de muerte súbita18. Además, el mantenimiento en el tiempo de episodios de hipercapnia e hipoxemia durante sueño y/o vigilia puede suponer mayor deterioro neurocognitivo en estos niños. Aunque la principal hipótesis para explicar la alteración respiratoria es la disfunción del centro respiratorio y sus quimiorreceptores, hay muy poca literatura al respecto17. En el estudio de Carrol et al.17 los pacientes con ROHHAD, en comparación con los diagnosticados de CCHS, muestran una sensibilidad periférica a la hipoxia e impulso quimioceptivo al oxígeno preservados, aunque sus hallazgos muestran importante susceptibilidad a la hipoxemia (a diferencia del CCHS, donde el problema principal es la hipercapnia). No son capaces, por tanto, de explicar la incapacidad de los pacientes con ROHHAD para mantener de forma espontánea normoxigenación y normoventilación, tal y como está ampliamente documentado en la literatura previa4. A pesar de ello, estipulan que quizá sea la conjunción de la ausencia de sensación de asfixia, con los efectos secundarios de la obesidad (posible descenso en el volumen residual funcional, volumen tidal y enfermedad pulmonar restrictiva), junto con el descenso del volumen tidal y del impulso inspiratorio ante situaciones de hipoxia hipercápnica. En definitiva, aunque no esté explicada su fisiopatología con detalle, sí es de suma importancia comprender que la falta de percepción y respuesta fisiológica normal en situaciones de hipoxemia o hipercapnia críticas hace que sea imprescindible un manejo respiratorio agresivo para intentar evitar episodios de muerte súbita y mayor déficit neurocognitivo. Así, el soporte respiratorio forma parte de los principales pilares del tratamiento de soporte. En función de las características de cada paciente, precisarán desde ventilación no invasiva nocturna hasta ventilación mecánica continua a través de traqueotomía (50% de pacientes según revisiones)12,17,18. El síndrome ROHHAD puede asociar disautonomía de diferentes tipos y gravedad. Lo más frecuente es la afectación oftalmológica, presentando alteración del reflejo fotomotor, alteraciones de la termorregulación (hipo e hipertermia), dismotilidad intestinal (estreñimiento o diarrea crónica) y alteraciones de la sensibilidad al dolor. También es importante recordar el rastreo de tumores de cresta neural asociados en hasta un 40% de los casos (ganglioneuromas y ganglioneuroblastomas), aunque al parecer no se ha descrito mejoría general del síndrome tras su exéresis4,6,15. Además, hay que tener en cuenta que el momento de presentación de estos varía mucho de unos pacientes a otros, puesto que incluso se han detectado más de 10 años después del inicio de los síntomas. Todo ello parece ir en contra de la hipótesis de su posible etiología paraneoplásica, si bien es cierto que no es posible afirmar que los tumores no pudiesen estar presentes previamente, siendo su crecimiento muy lento, o incluso pudiendo presentar regresión espontánea en aquellos pacientes en los que no se detectan.
La información disponible en la actualidad en cuanto a la esperanza de vida y el pronóstico a largo plazo de estos pacientes es limitada, puesto que no hay casos descritos que hayan alcanzado la edad adulta18.
Respecto al tratamiento, la literatura refleja la experiencia de una paciente con ROHHAD tras tratamiento con inmunoglobulina por vía IV, que mejoró la conducta y la adipsia14, pero sin modificar su disfunción endocrina. En nuestra paciente este fue el tratamiento inicial, que sí permitió un buen control electrolítico (no presentó alteraciones graves del sodio durante este período) y de la temperatura, pero persistía adipsia y sobre todo el trastorno de conducta (auto y heteroagresividad), refractario a neurolépticos. Dada la afectación en la calidad de vida que presentaba, ofrecimos una alternativa a la familia. Se decidió entonces iniciar la pauta de ciclofosfamida a 750mg/m2/dosis mensual, que fue solo parcialmente efectiva (la mejoría de la conducta duraba 2-3 días) y seguimos administrando inmunoglobulinas de modo mensual (2g/kg IV). La experiencia de Paz-Priel et al.7 en una paciente publicada y 2 más no publicados nos animaron a utilizar ciclofosfamida a dosis elevada (50 mg/kg/día durante 4 días consecutivos), con respuesta muy satisfactoria en nuestra paciente, aunque la inmunosupresión posterior precipitó un cuadro de shock séptico grave y múltiples complicaciones a lo largo de su prolongado ingreso en cuidados intensivos. Tras su recuperación, la conducta mejoró significativamente, pero persistían importantes dificultades para mantener unos niveles de sodio adecuados, apreciándose frecuentes episodios de hipo/hipernatremia ante desencadenantes como infecciones. Por este motivo, se siguieron administrando inmunoglobulinas mensualmente (2g/kg), ya que lo relacionamos con menor probabilidad de descompensaciones hidroelectrolíticas y mejor regulación térmica. Otros autores han publicado su experiencia con otros fármacos inmunosupresores como rituximab (anticuerpo monoclonal anti-CD20, a dosis de 375mg/m2 durante 6 semanas), apreciando mejoría transitoria en la termorregulación e hipoventilación, con reaparición de los síntomas al poco tiempo7,15. De todos modos, incluso en los casos que responden al tratamiento, se aprecia algún grado de secuela cognitiva7. Por último, cabe mencionar algunas consideraciones anestésicas de importancia, puesto que los pacientes con síndrome ROHHAD pueden requerir varias intervenciones quirúrgicas y, como hemos visto, su manejo puede ser complejo. Como el diagnóstico es clínico y por exclusión, puede hacerse de forma tardía, por lo que se pueden encontrar pacientes no diagnosticados en el momento de la cirugía, que presenten después un destete complicado del respirador. En aquellos casos con diagnóstico de ROHHAD establecido se deberá tener en cuenta la posible gastroparesia (con posibilidad de residuo alimentario a pesar de tiempo de ayunas adecuado), las alteraciones hidroelectrolíticas, autonómicas y la afectación neurológica. En general, y según opiniones publicadas, se prefiere el uso de anestésicos de acción corta y con mínima afectación respiratoria, junto con asociación de agentes ansiolíticos. Además, se recomienda vigilancia estrecha en el postoperatorio inmediato por el alto riesgo de complicaciones19.
Consideramos que el diagnóstico precoz de ROHHAD es fundamental para iniciar el tratamiento adecuado de los trastornos endocrinológicos y el seguimiento estrecho multidisciplinar, prestando especial atención a la aparición de hipoventilación alveolar central y su necesidad de soporte respiratorio. La alta incidencia de episodios de parada cardiorrespiratoria domiciliaria y su elevada mortalidad son los responsables del pronóstico poco esperanzador de esta entidad. Dados su heterogenicidad y los síntomas comunes con otros síndromes, es fundamental conocer esta patología tan poco habitual y mantener un alto índice de sospecha.
FinanciaciónNo se han utilizado.
Conflicto de interesesNo existen conflictos de interés.

