



ROHHAD syndrome (rapid-onset obesity with hypothalamic dysregulation, hypoventilation, and autonomic dysregulation) is a rare and complex disease, presenting in previously healthy children at the age of 2-4 years. Up to 40% of cases are associated with neural crest tumours.
DevelopmentWe present the case of a 2-year-old girl with symptoms of rapidly progressing obesity, who a few months later developed hypothalamic dysfunction with severe electrolyte imbalance, behaviour disorder, hypoventilation, and severe autonomic dysregulation, among other symptoms. Although the pathophysiology of this syndrome remains unclear, an autoimmune hypothesis has been proposed for ROHHAD. Therefore, after obtaining a limited response to intravenous immunoglobulins, we decided to test the response to a high dose cyclophosphamide (low dose was not effective either). Unfortunately our patient experienced many severe complications (among them central pontine myelinolysis, from which the patient recovered, and failure to wean from the ventilator requiring tracheostomy and long term ventilation) that required a prolonged ICU stay. Although her behaviour improved, our patient unfortunately died suddenly at home at the age of 5 due to respiratory pathology.
ConclusionsROHHAD syndrome is a rare and little-known disease which requires a multidisciplinary approach because it involves complex symptoms and multiple organ system involvement. Alveolar hypoventilation should be identified early and appropriate treatment should be started promptly for the best possible outcome. Immunomodulatory treatment with immunoglobulins, cyclophosphamide, or rituximab has previously resulted in symptom improvement in some cases. Because of the low incidence of the syndrome, multi-centre studies must be carried out in order to gather more accurate information about ROHHAD pathophysiology and design an appropriate therapeutic approach.
El síndrome obesidad de rápida progresión, disfunción hipotalámica, hipoventilación alveolar y disregulación autonómica (ROHHAD) es una entidad infrecuente y compleja con comienzo en niños sanos a los 2-4 años, donde además un 40% se relaciona con tumores de la cresta neural.
DesarrolloPresentamos el caso de una niña que comenzó a los 2 años con un cuadro de obesidad de rápida progresión y posteriormente asoció disfunción hipotalámica con trastornos electrolíticos graves, trastorno de conducta, hipoventilación y disautonomía graves, entre otros. Aunque su fisiopatología no está aclarada, una de las hipótesis actuales del ROHHAD es autoinmune y, tras respuesta limitada a inmunoglobulinas por vía intravenosa, se decidió probar respuesta a ciclofosfamida a dosis altas (a dosis bajas tampoco fue eficaz). Esto motivó múltiples complicaciones graves posteriores que requirieron estancia prolongada en la UCI (a destacar mielinólisis central pontina recuperada e imposibilidad de destete del respirador, requiriendo traqueotomía para continuar asistencia respiratoria). Aunque la conducta mejoró, el desenlace fue fatal a los 5 años debido a un episodio de muerte súbita en domicilio por su patología respiratoria.
ConclusionesSe trata de una patología poco conocida que precisa un abordaje multidisciplinar, dada la complejidad de los síntomas y su implicación multisistémica. Es necesario identificar precozmente la hipoventilación alveolar y el inicio de tratamiento adecuado por su implicación pronóstica. La experiencia con inmunomoduladores como inmunoglobulinas, ciclofosfamida o rituximab muestra una mejoría de los síntomas en algunos casos. Sería deseable realizar estudios multicéntricos, dada la baja incidencia del síndrome, para esclarecer su fisiopatología y diseñar su adecuado abordaje terapéutico.
Rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation, (ROHHAD syndrome) is a rare disorder associated with alterations in the endocrine system (mainly the hypothalamus), the autonomic nervous system, and the respiratory system (central hypoventilation). To date, fewer than 100 cases of ROHHAD syndrome have been described worldwide. Although the case of a Spanish patient potentially meeting current diagnostic criteria for ROHHAD syndrome was published in 1996,1 ours is the first confirmed case of ROHHAD syndrome to be reported in Spain.
For many years, ROHHAD syndrome has overlapped with and may have been mistaken for late-onset congenital central hypoventilation syndrome (LO-CCHS). The description of mutations in the PHOX2B gene, which are present in patients with CCHS2,3 and absent in those with ROHHAD,4–6 enabled a distinction to be made between these 2 nosological entities.
The term ROHHAD and the diagnostic criteria for the syndrome were proposed by Ize-Ludlow et al.4 The initial symptom of ROHHAD is usually hyperphagia, occurring in previously healthy children aged 2 to 4 years. Hyperphagia results in dramatic weight gain in a short period of time (10-15kg in 6-12 months). In the following months and years, patients begin to display other alterations in hypothalamic function, the most frequent being electrolyte imbalance and dysautonomia.
These patients also experience early-onset obstructive sleep apnoea and subsequently central alveolar hypoventilation; prognosis of ROHHAD largely depends on how the latter is managed. Behavioural disorders and cognitive and speech impairment are very common symptoms. Around 40% of the cases published also present tumours of neural crest origin (ganglioneuroma, ganglioneuroblastoma) throughout the course of the disease; on some occasions, these tumours may even appear several years after symptom onset.4–6 This explains why this syndrome is currently known as ROHHAD-NET (neuroectodermal tumours).
We present our experience with the first case of ROHHAD to be published in Spain and summarise the results of our literature review.
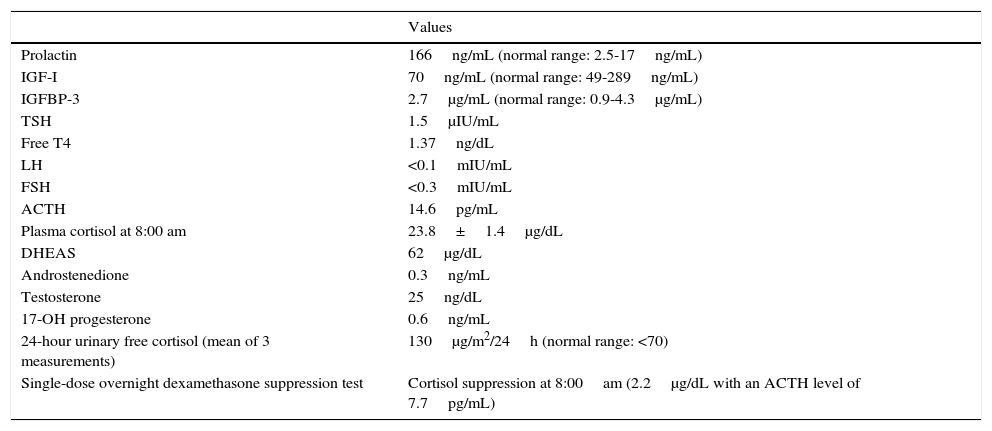
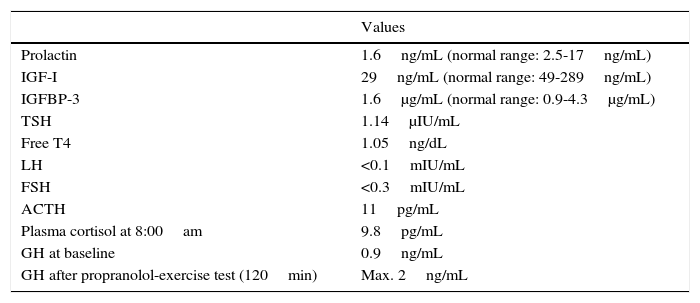
Clinical caseOur patient was referred from primary care at the age of 2 years and 10 months due to rapid-onset obesity (8kg in 10 months) and increased appetite. Her mother had experienced no complications during pregnancy or delivery, and the neonatal period had been normal, with the patient displaying normal growth and psychomotor development. She weighed +3.8 SD and was −0.8SD tall, in line with her target height (−0.5SD). She displayed a normal growth velocity and had a BMI of +6.8SD. The examination detected no alterations, with no cushingoid features or acanthosis nigricans; blood pressure was normal. She was the first child born to young, healthy, non-consanguineous parents. The patient underwent a series of laboratory analyses. Biochemical studies of blood and urine yielded normal results. A hormonal analysis detected severe, persistent hyperprolactinaemia (166ng/mL; normal range: 2.5–17ng/mL), suggestive of pituitary adenoma or hypothalamic dysfunction. The results of these hormonal tests are summarised in Table 1. Primary hypercortisolism was ruled out. Given the persistent hyperprolactinaemia and high urine cortisol levels, the study of central hypercortisolism included a contrast-enhanced MRI scan of the brain and the hypothalamus and pituitary gland; no abnormalities were detected. Bilateral convergent strabismus appeared 2 months later; the results of the eye fundus, cranial nerve, and neurological examinations were all normal. The patient's behaviour considerably worsened shortly afterwards, with episodes of self- and hetero-aggression, hyperactivity, and impulsivity, which were refractory to neuroleptics (risperidone, aripiprazole, and olanzapine). The patient's low sensitivity to pain was noteworthy: she hit herself hard in the head without complaining and even smiled during venipunctures. A chest and abdomen MRI scan revealed no anomalies, and genetic studies for Prader–Willi syndrome and Smith–Magenis syndrome yielded negative results. When the patient was 3 years and 3 months old, she experienced the first episode of electrolyte imbalance with severe hypernatraemia (175mEq/L), with no findings suggestive of diabetes insipidus (DI). She was admitted to hospital on multiple occasions due to episodes of hyper- and hyponatraemia; some of these episodes coincided with infections, while others had no apparent cause. The patient's parents were instructed to control the child's fluid intake. Laboratory analysis results did not clearly indicate either deficient or excessive secretion of antidiuretic hormone (ADH). During one of her admissions to hospital, the patient experienced episodes of apnoea-hypopnoea, mainly during sleep; according to her parents, she had displayed cyanosis on several occasions while at home. Furthermore, the patient began to display signs of dysautonomia: episodes of hypothermia (down to 32°C) with no apparent cause, a tendency to bradycardia, and intestinal motility disorders (severe gastro-oesophageal reflux disorder beginning at age 3, requiring Nissen fundoplication, and complete rectal prolapse requiring rectopexy). The patient's weight, which had increased very rapidly at first, normalised at the age of 3 due to feeding difficulties. ROHHAD syndrome was suspected based on the patient's progression and the presence of the signs and symptoms previously described. The patient tested negative for serum and CSF autoantibodies (including anti-NMDA, anti-AMPA, and anti-GABAB). Results from a study of inborn errors of metabolism (amino acids, organic acids, SAICAR, sulfite oxidase, lactate/pyruvate, and neurotransmitters in CSF) were normal. Karyotyping and molecular study of the PHOXB2 gene revealed no abnormalities. At the time of diagnosis, we decided to start treatment with intravenous (IV) immunoglobulins dosed at 2g/kg/month for 8 months; behavioural disorders did not improve during that period, but no further episodes of severe hyper- or hyponatraemia or hypothermia were recorded. Regarding endocrinological findings, from the age of 4 the patient displayed delayed growth (height −2 SD, weight −0.2 SD, BMI +1.2SD, and GV −3.5SD) and was diagnosed with growth hormone deficiency. Table 2 summarises our patient's hormone levels at the age of 4. At the age of 5, she displayed central hypothyroidism (TSH 0.19, fT4 0.2ng/dL); we decided to start replacement therapy with thyroxine dosed at 3μg/kg/day. As a result of this treatment, IGF-I and IGFBP-3 levels normalised (52ng/mL and 1500μg/mL, respectively). The patient subsequently displayed chronic central adrenal insufficiency (baseline cortisol 1.3μg/dL with borderline response in the ACTH test, 18μg/dL at 60minutes from administration of 250μg of synthetic ACTH), starting treatment with oral hydrocortisone dosed at 13mg/m2/day. Treatment improved bradycardia, somnolence, hypothermia, and the tendency to mild hypoglycaemia; behavioural disorders persisted. As new symptoms appeared, and given the suspicion of an autoimmune origin of the disease, we started treatment with cyclophosphamide dosed at 750mg/m2/month for 4 months, leading to mild, transient improvements after each cycle. We then decided on high-dose cyclophosphamide treatment based on the experience of Paz-Priel et al.7 with a published case and 2 further unpublished cases of patients experiencing behavioural improvements after administration of high-dose cyclophosphamide (50mg/kg/day for 4 consecutive days). Coinciding with the neutropaenia induced by cyclophosphamide treatment, the patient worsened suddenly and developed symptoms of sepsis of respiratory origin due to Pseudomonas aeruginosa; she was admitted to the paediatric intensive care unit and remained in hospital for nearly 4 months due to multiple complications. In addition to haemodynamic instability due to septic shock, she displayed symptoms of dysautonomia, including bradycardia and hypothermia, complicating the existing septic shock symptoms. We started treatment with corticosteroids since septic shock was refractory to catecholamines (hydrocortisone dosed at 100mg/m2). After slow recovery from bone marrow failure, acute symptoms resolved; however, the patient was continued on mechanical ventilation after multiple attempts at extubation (massive atelectasis and persistence of central sleep apnoea). Furthermore, she displayed symptoms of spastic tetraplegia, involvement of the lower cranial nerves, and low level of consciousness; a brain MRI scan revealed signs of central pontine myelinolysis. During hospitalisation, the patient had experienced multiple episodes of electrolyte imbalance, which were very difficult to manage and attributable to hypothalamic dysfunction. She experienced episodes of DI, especially during the phase of shock, alternating with periods of syndrome of inappropriate antidiuretic hormone secretion (SIADH), the latter being more frequent. During episodes of DI, serum sodium levels reached 175mEq/L, requiring desmopressin and strict serum sodium control. During the episodes of SIADH, fluid intake was restricted and diuretics were administered, achieving a decrease in serum sodium levels (123mg/dL). After the acute process, the physicians’ general impression was that a more “physiological” management was most suitable for the patient: when possible, they avoided IV fluid therapy, especially with hypotonic solutions, and enteral nutrition and fluid administration to prevent hyponatraemia. Although no abrupt changes in sodium levels or osmolarity were detected, the patient's baseline hypothalamic dysfunction may have predisposed to central pontine myelinolysis during the acute process. After reviewing the literature available on the topic,8,9 we decided to administer IV immunoglobulins (0.4g/kg for 5 days). Treatment led to significant improvements over the following weeks: the patient's level of consciousness improved progressively and she displayed spontaneous mobility of all 4 limbs, started articulating speech, and was able to chew, swallow, and even walk. We decided to perform a tracheostomy to provide respiratory support. The patient was ultimately discharged and followed up by home care services, and she required multiple specialist consultations. Dysautonomia progressed; the patient underwent pacemaker implantation due to severe sinus bradycardia at 6 months from discharge. Interestingly, she presented several infectious processes but rarely developed fever due to her tendency to hypothermia (baseline temperature 35°C).
Hormone levels at baseline (age: 2 years and 10 months).
| Values | |
|---|---|
| Prolactin | 166ng/mL (normal range: 2.5-17ng/mL) |
| IGF-I | 70ng/mL (normal range: 49-289ng/mL) |
| IGFBP-3 | 2.7μg/mL (normal range: 0.9-4.3μg/mL) |
| TSH | 1.5μIU/mL |
| Free T4 | 1.37ng/dL |
| LH | <0.1mIU/mL |
| FSH | <0.3mIU/mL |
| ACTH | 14.6pg/mL |
| Plasma cortisol at 8:00 am | 23.8±1.4μg/dL |
| DHEAS | 62μg/dL |
| Androstenedione | 0.3ng/mL |
| Testosterone | 25ng/dL |
| 17-OH progesterone | 0.6ng/mL |
| 24-hour urinary free cortisol (mean of 3 measurements) | 130μg/m2/24h (normal range: <70) |
| Single-dose overnight dexamethasone suppression test | Cortisol suppression at 8:00am (2.2μg/dL with an ACTH level of 7.7pg/mL) |
Hormone levels at the time of diagnosis of ROHHAD (age: 4 years).
| Values | |
|---|---|
| Prolactin | 1.6ng/mL (normal range: 2.5-17ng/mL) |
| IGF-I | 29ng/mL (normal range: 49-289ng/mL) |
| IGFBP-3 | 1.6μg/mL (normal range: 0.9-4.3μg/mL) |
| TSH | 1.14μIU/mL |
| Free T4 | 1.05ng/dL |
| LH | <0.1mIU/mL |
| FSH | <0.3mIU/mL |
| ACTH | 11pg/mL |
| Plasma cortisol at 8:00am | 9.8pg/mL |
| GH at baseline | 0.9ng/mL |
| GH after propranolol-exercise test (120min) | Max. 2ng/mL |
Approximately 9 months after treatment with cyclophosphamide, the patient attended school and completed tasks independently (before treatment she was completely dependent on her carers), controlled her sphincters, ate without assistance, walked independently, played with other children, recovered sensitivity to pain, and was able to hold simple, coherent conversations. Although the patient continued to display mild cognitive impairment and presented motor sequelae (mild pyramidal signs), her behaviour improved considerably (requiring only low doses of risperidone), resulting in better quality of life for the patient and her family. The patient continued to be followed up by the oncology department for tumour screening and required intermittent oxygen therapy and mechanical ventilation through a tracheostomy due to central sleep apnoea and alveolar hypoventilation during sleep. Respiratory problems constitute the main predictor of poor prognosis of ROHHAD syndrome. Unfortunately, at the age of 6 our patient died suddenly while at home; she was not receiving mechanical ventilation at the time of death, and the pacemaker functioned correctly, according to the emergency services. The parents did not consent to autopsy.
DiscussionROHHAD, an extremely complex entity requiring a high level of suspicion for diagnosis, must be managed by a multidisciplinary team including endocrinologists, psychiatrists, surgeons, pneumologists, oncologists, neuropaediatricians, and cardiologists, among other specialists. Early management, even in the case of conservative treatment, is essential to improve prognosis and prevent complications. The first case of ROHHAD was described in 1965 by Fishman et al.10 In addition to the typical phenotype of the syndrome, the patient experienced late-onset central hypoventilation and hypothalamic dysfunction. It was not until 2000, however, that Katz et al.11 established a clear distinction between LO-CCHS and late-onset central hypoventilation syndrome with hypothalamic dysfunction (LO-CHD/HD), describing the case of a patient with LO-CHD/HD and reviewing an additional 10 cases published in the literature. These authors proposed a list of common signs and symptoms of this condition, which is still used at present. In 2007, Ize-Ludlow et al.4 found no mutations in the PHOX2B gene in these patients and coined the term ROHHAD. The new term ROHHAD-NET, however, refers to the frequent association between ROHHAD and this type of tumour.6 We prefer the classic term ROHHAD, given that ROHHAD-NET is much less widely used. Diagnosis of ROHHAD is clinical and must be made by exclusion, given that the aetiology of the syndrome is yet to be determined.12 ROHHAD probably results from a combination of genetic predisposition and paraneoplastic or immunological factors. However, no epigenetic changes or genetic alterations specific to this syndrome have been identified, and there is no evidence of its association with any autoimmune process. The hypothesis that ROHHAD may be associated with paraneoplastic or autoimmune factors was proposed in 1995.13 This theory should, however, be considered with caution, since the experience with immunomodulatory or immunosuppressive treatment published to date corresponds to individual cases or case series.7,14,15 The literature describes some cases of lymphocytic and histiocytic infiltration of the hypothalamus,13 and an association between ROHHAD and presence in the CSF of such biomarkers as oligoclonal bands has been suggested in recent years.16 Furthermore, the publication of a case of ROHHAD in only one of a pair of monozygotic twins12 has raised doubts over the genetic origin of the syndrome. Although this may point to the involvement of modifier genes, it also supports the hypothesis that ROHHAD is caused by other, non-genetic factors. Clinical diagnostic criteria for ROHHAD must include rapid weight gain and hypoventilation appearing after the age of one and a half years. Patients must also present hypothalamic dysfunction plus at least one of the following disorders: rapid-onset obesity, hyperprolactinaemia, central hypothyroidism, electrolyte imbalances, failed growth hormone stimulation test, corticotrophin deficiency, or altered onset of puberty.
To confirm diagnosis, we must also rule out CCHS and Prader–Willi syndrome.15 As previously described, children with ROHHAD show normal growth and development until symptom onset, which takes place between the ages of 1.5 and 9 years. Symptoms progress over the course of several years. Weight gain, the most common initial manifestation, is followed by hypothalamic dysfunction,4 as in our case. The progression of endocrine symptoms in our patient is noteworthy: she initially presented hyperprolactinaemia and a pattern suggestive of pseudo-Cushing syndrome, but symptoms evolved to generalised hypothalamic and pituitary gland dysfunction (central hypothyroidism and adrenal insufficiency, and low levels of IGF-I, prolactin, FSH, and LH). Furthermore, she exhibited the hormonal changes previously described in patients with this syndrome, such as electrolyte imbalances (episodes of hyper- and hyponatraemia), although laboratory analysis did not clearly reveal dysfunction of ADH secretion. Unlike in the cases previously reported, our patient's body weight progressed satisfactorily; she had a BMI of +0.14SD in her last follow-up assessment, which was probably linked to the severity of gastro-oesophageal reflux, multiple complications, and long hospital stays. Delayed growth (height −3.38 SD), however, may have normalised progressively after initiating treatment for central hypothyroidism. Respiratory problems associated with ROHHAD always arise at some point during the disease's progression.1,4,15,17 Patients initially present obstructive sleep apnoea and subsequently develop central alveolar hypoventilation. The latter is characterised by the absence of signs of respiratory difficulty associated with hypercapnia and/or hypoxaemia, leading to progressive inadequate ventilation; this, combined with dysautonomia, entails a high risk of sudden death.18 Furthermore, repeated episodes of hypercapnia and hypoxaemia during sleep and/or wakefulness may result in more severe neurocognitive impairment in these children. Although dysfunction of the respiratory centres and their chemoreceptors is the most likely explanation for respiratory problems, very little has been published on this topic.17 In the study by Carroll et al.17, peripheral sensitivity to hypoxia and chemoreceptive response to oxygen were similar in patients with ROHHAD and those diagnosed with CCHS. However, the former also showed significant susceptibility to hypoxaemia, unlike those with CCHS, who mainly experienced hypercapnia. Therefore, these authors could not explain the inability of ROHHAD patients to maintain normal oxygenation and ventilation spontaneously, as has been extensively reported in the literature.4 However, they hypothesise that this may be due to a combination of factors, including the absence of a sensation of asphyxia, the secondary effects of obesity (possible decrease in functional residual capacity, tidal volume, and restrictive lung disease), and decreased tidal volume and inspiratory impulses in situations of hypercapnic hypoxia. In summary, although the exact pathophysiological mechanisms of ROHHAD are yet to be determined, it is of utmost importance to be aware that a lack of perception and normal physiological response in situations of severe hypoxaemia or hypercapnia makes it impossible to provide aggressive respiratory therapy to prevent more severe neurocognitive deficits and sudden death. Ventilatory support therefore constitutes one of the main pillars of treatment for these patients. Each patient has specific respiratory needs, ranging from nocturnal non-invasive ventilation to continuous mechanical ventilation through tracheostomy (50% of patients, according to the literature).12,17,18 The type and the degree of severity of dysautonomia in ROHHAD syndrome varies from case to case. These patients most frequently present eye involvement, with pupillary light reflex alterations, impaired thermoregulation (hypo- and hyperthermia), intestinal dysmotility (constipation or chronic diarrhoea), and altered pain sensitivity. Patients with ROHHAD should also be screened for tumours of neural crest origin, present in around 40% of cases (ganglioneuroma and ganglioneuroblastoma); however, no overall improvement of the disease has been described after tumour resection.4,6,15 We should also bear in mind that these tumours may present at different stages of the disease, in some cases even 10 years after symptom onset. The above seems to contradict the hypothesis that ROHHAD is of paraneoplastic origin. However, we cannot be certain that these tumours were not present long before they were detected (they may have grown at a very slow pace). Likewise, spontaneous regression may explain why no tumours are detected in some patients.
Current data on the life expectancy and long-term outcomes of patients with ROHHAD is limited, since no cases have been reported in which patients reached adulthood.18
Regarding treatment, the literature describes the case of a patient with ROHHAD in whom IV immunoglobulins improved behaviour and adipsia but failed to resolve endocrine dysfunction.14 Our patient was initially treated with IV immunoglobulins, achieving good electrolyte balance (no severe sodium level alterations were recorded during that period) and temperature control; however, treatment failed to control adipsia and particularly behavioural disorders (self- and hetero-aggression), which were refractory to neuroleptics. In view of the patient's and her family's poor quality of life, we decided to start treatment with cyclophosphamide dosed at 750mg/m2/month. Treatment was only partially effective (behaviour improvements lasted 2-3 days), and IV immunoglobulins were continued at monthly doses of 2g/kg. High-dose cyclophosphamide (50mg/kg/day for 4 consecutive days) was administered following the experience reported by Paz-Priel et al.7, with one published case and 2 unpublished cases. The patient responded excellently, although subsequent immunosuppression led to severe septic shock and multiple complications throughout her long stay at the paediatric intensive care unit. Behavioural disorders improved considerably after her recovery, although sodium levels continued to be outside the normal range, with frequent episodes of hypo- and hypernatraemia in the event of such trigger factors as infections. The patient continued receiving immunoglobulins at a monthly dose of 2g/kg since this treatment seemed to be associated with better thermoregulation and lower risk of electrolyte imbalances. Several authors have published their experience with other immunosuppressants, such as rituximab (anti-CD20 monoclonal antibody, dosed at 375mg/m2 for 6 weeks), noting transient improvements in thermoregulation and hypoventilation (symptoms reappeared shortly thereafter).7,15 However, even those patients achieving good treatment response present cognitive sequelae to some extent.7 Lastly, we would make some remarks on anaesthetic management, given that patients with ROHHAD may need to undergo surgery on several occasions; as shown previously, surgical management of these patients is complex. Given that diagnosis of ROHHAD is clinical and must be made by exclusion, some patients may have not been diagnosed with the condition at the time of surgery; these patients may be difficult to wean from mechanical ventilation. In patients with a definite diagnosis of ROHHAD, doctors should consider the possibility of gastroparesis (with presence of food residue in the stomach despite adequate fasting time), electrolyte imbalances, autonomic dysfunction, and neurological impairment. In general terms, and according to published opinion, short-acting anaesthetics with minimal respiratory effects, combined with anxiolytics, are the anaesthetic approach of choice. Furthermore, close post-operative follow-up is recommended due to the high risk of complications.19
Early diagnosis of ROHHAD is essential for appropriate management of endocrine dysfunction and close multidisciplinary follow-up. Special attention should be paid to onset of central alveolar hypoventilation due to the need for respiratory support. The high frequency of episodes of cardiac arrest at home and the associated high mortality rate are responsible for the poor prognosis of ROHHAD. Symptoms of ROHHAD are heterogeneous and may overlap with those of other conditions. Gaining a deeper understanding of this syndrome and maintaining a high level of suspicion is therefore essential.
FundingThis study has received no funding of any kind.
Conflicts of interestThere are no conflicts of interest to declare.
Please cite this article as: Ibáñez-Micó S, Marcos Oltra AM, de Murcia Lemauviel S, Ruiz Pruneda R, Martínez Ferrández C, Domingo Jiménez R. Síndrome ROHHAD (obesidad de rápida progresión, disfunción hipotalámica, hipoventilación y disregulación autonómica). Presentación de un caso y revisión de la literatura. Neurología. 2017;32:616–622.
