



Hereditary spastic paraplegia refers to a group of neurodegenerative diseases presenting great heterogeneity both in clinical and genetic terms, which makes their diagnosis a challenging process. The true prevalence of hereditary spastic paraplegia in Spain is currently unknown; however, a recent study has estimated a rate of 2.24 cases per 100 000 population. In Spain, the most frequent autosomal dominant hereditary spastic paraplegias are SPG4 and SPG17, and SPG7 and SPG11 are the most frequent autosomal recessive forms.1 Autosomal dominant, autosomal recessive, X-linked, and mitochondrial forms have been identified.
At least 86 genetic types have been described. There are pure and complex forms, which are accompanied by neurological or non-neurological symptoms. The first classification was established by Harding in 1983.2
Advances in the field of genomics represent a revolution in this disease, contributing to their diagnosis and the discovery of new variants.3 We present the clinical case of a patient diagnosed with SPG46, a recessive form caused by a variant of the gene encoding the beta-glucosidase 2 enzyme (GBA2).
Our patient, a 57-year-old white woman, was referred to the neurology department due to a gait disorder progressing for several years. She presented no family history of neurodegenerative diseases, and her parents were non-consanguineous. The first motor symptoms (difficulty running) manifested during childhood, accompanied by poor performance at school. She was diagnosed with mild intellectual disability. Motor symptoms progressed slowly, becoming more apparent in the fifth decade of life, when she presented frequent falls. By the age of 54, she required a wheelchair.
The neurological examination revealed a Mini–Mental State Examination score of 26/30. The patient presented hypometric horizontal saccades, without nystagmus. Motor symptoms included dystonia of the hands, predominantly distal spastic tetraplegia, mainly affecting the lower limbs (proximal and distal muscle strength 4/5 in all 4 limbs), pyramidal signs (overactive tendon reflexes, bilateral extensor plantar reflex, and bilateral Achilles clonus), and pes cavus and equinus. Sensory symptoms included impaired positional sensitivity in the lower limbs. These symptoms were accompanied by such cerebellar symptoms as bradylalia, dysarthria, bilateral dysmetria on the finger-to-nose test and heel-to-knee test, and ataxic-spastic gait. An examination by the ophthalmology department diagnosed bilateral cataracts. A complete blood analysis (including thyroid hormones and vitamins, lactic and pyruvic acid, copper and ceruloplasmin, muscle enzymes, amino acids, sterols, and branched chain fatty acids) revealed no remarkable findings. The cerebrospinal fluid (CSF) study revealed elevated protein levels (77.9 mg/dL, normal range: 15–45) together with intrathecal IgG synthesis (oligoclonal in CSF and polyclonal in serum) with CSF IgG values of 4.75 mg/dL (normal range: 1–4), an IgG index of 0.00036, and a CSF albumin level of 27.7 mg/dL (normal range: 13.9–24.6).
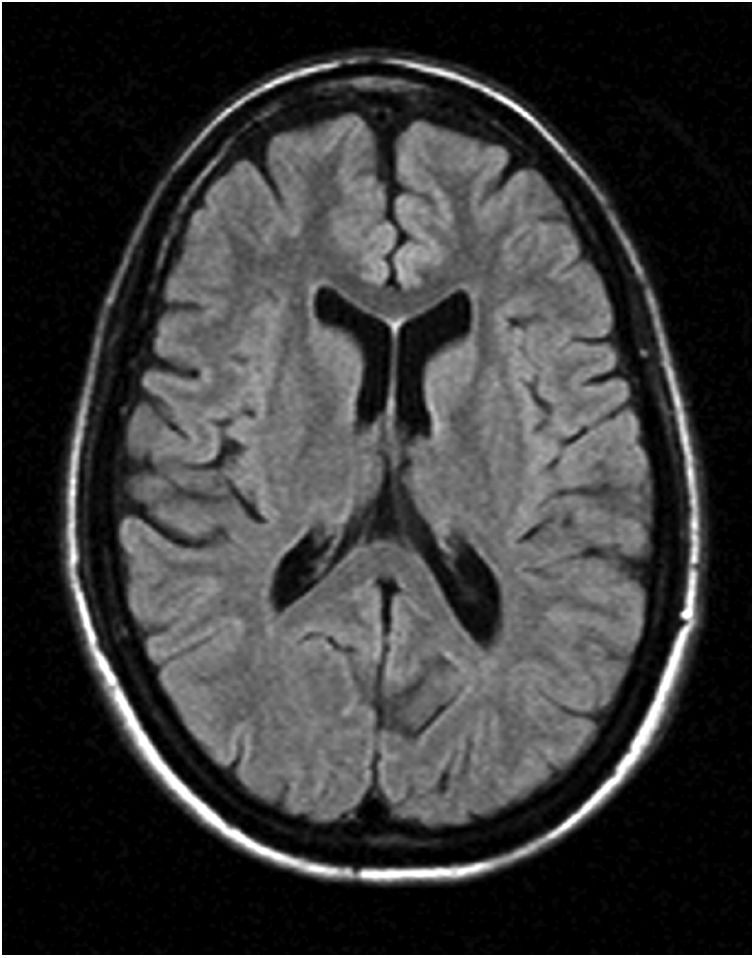
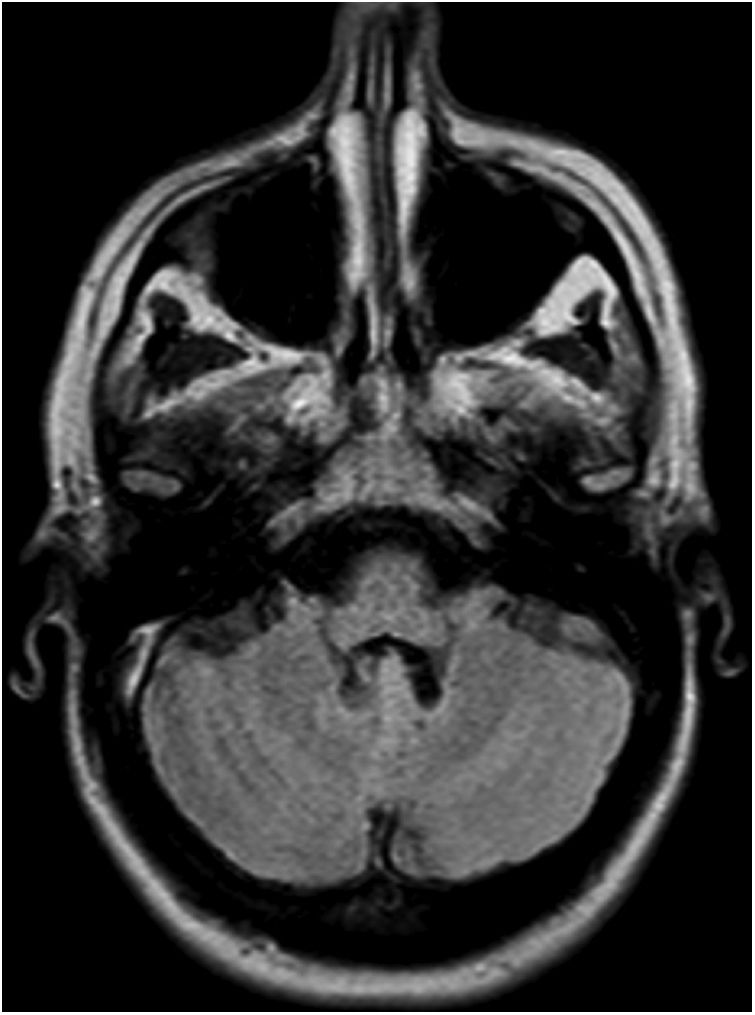
Somatosensory evoked potentials in the lower limbs showed increased latencies bilaterally, suggesting demyelination. A peripheral nerve study identified mixed (motor and sensory), mainly demyelinating neuropathy (probable secondary demyelination) of distal predominance. A brain (Fig. 1 and 2) and cervical spine MRI scan; a chest, abdomen, and pelvis CT scan; and a biopsy of the deltoid muscle and sural nerve all showed normal results. A polymerase chain reaction (PCR) study of spinocerebellar ataxias (SCA1, SCA2, SCA3, SCA6, SCA7, SCA12, SCA17, DRPLA) revealed no alterations.


Lastly, we conducted a genetic study of spastic paraplegias (NPC1, NPC2, GBA, GBA2, PSAP, POLG, APTX, HFE, CYP27A1, and ATP7A genes), taking into account the genotype-phenotype correlation, using massive DNA sequencing (AmpliSeq and ultrasequencing with the Ion PGM platform). The study revealed the homozygous deletion of 2 nucleotides in exon 9 of the GBA2 gene: Nm_020944:exon9:c.1475_1476del, predicted to result in a frameshift from codon 492 (NP_065995.1:p.T492fs). This variant was not found in a search of population and pathogenic variant databases.
Furthermore, a genetic study of the patient’s brother (asymptomatic) revealed that he was a heterozygous carrier of the variant. No family segregation studies were performed (the parents and grandparents were deceased, and her only brother had no children). Results were confirmed by PCR and bidirectional Sanger sequencing, and a diagnosis of SPG was established.
The GBA2 gene is located on chromosome 9, and has 17 exons.4 It encodes the beta-glucosidase 2 enzyme, which catalyses the conversion of glucosylceramide to free glucose and ceramide.5 Glucosylceramide is a precursor of sphingolipids, which are abundantly found in the cell membranes of the central nervous system. They have been observed to increase in number throughout growth and to change with axonal development and differentiation, and are important for the development of these neuronal structures.6 The first cases of GBA2 variants were described in 2013, in 3 families from Tunisia.7 Since then, patients with GBA2 variants have been described in Italy, China, Japan, Germany, and India,8–13 both with the classic phenotype of pyramidal signs in the lower limbs accompanied by cerebellar symptoms and progressive cognitive impairment, and with such other symptoms as dystonia.12,13
In conclusion, we present the first case of SPG46 described to date in Spain. An unusual feature in our patient is the elevated CSF protein level, which has not been described in the reviewed literature. Advances in genomic sequencing have led to a revolution in hereditary spastic paraplegias. We would underscore the relevance of continuing to implement methods for genomic diagnosis in the field of neurology, as we believe that this will lead to findings in diseases not currently linked to a specific gene, and will open new pathways for possible therapies.
FundingNone.
Conflicts of interestNone.
