



Periventricular nodular heterotopia (PNH) is a malformation of cortical development caused by impaired neuronal migration that prevents certain bundles of neurons from travelling from the marginal periventricular area to their final location in the cortex.1 This creates nodules of ectopic grey matter that lead to erroneous connections, focal deficits, and frequently refractory epilepsy.2
PNH typically manifests in adolescence and may be associated with a wide range of symptoms; approximately 90% of patients will present epilepsy and cognitive delay. Depending on the mutation, PNH may also cause malformations in other systems, especially the cardiovascular system.3
Its worldwide prevalence is unknown due to the rareness of the condition. The available data were obtained from case series, with one providing data on adult patients with epilepsy4; approximately 2% of these patients have PNH, which accounts for 20% of cases of malformations of cortical development.
Regarding the aetiology of the condition, studies have identified mutations in specific genes that are responsible for neuronal migration to the cortex.5 In particular, mutation of the FLNA gene, involved in the regulation of cell stability and motility through several biological systems, and which follows an X-linked dominant inheritance pattern,6 is associated with higher fetal and perinatal mortality in males, with better outcomes in female patients. We should mention that the reported cases are frequently sporadic.7,8
Diagnosis is established by brain MRI, which typically reveals confluent masses of ectopic grey matter that are usually bilateral, isointense, and distributed throughout the area surrounding the lateral ventricles.9 Furthermore, studies have also identified abnormalities in the corpus callosum, including hypoplasia, dysgenesis, and agenesis.10 Electroencephalography studies reveal epileptogenic discharges at the level of the periventricular nodules or in the underlying cortex, or both simultaneously.11
Regarding treatment, surgical resection may be recommended in patients presenting seizures with specific, localised focal onset or displaying resistance to pharmacological treatment, after exhaustive evaluation.12
We describe the case of a 22-year-old woman with history of epilepsy of unknown aetiology since the age of 15. She was receiving treatment with phenobarbital and carbamazepine at optimal doses, without achieving adequate seizure control. At the time of assessment, she was 11.2 weeks pregnant with a single fetus. Her family history included seizures (her mother) during adolescence, which resolved after her first pregnancy.
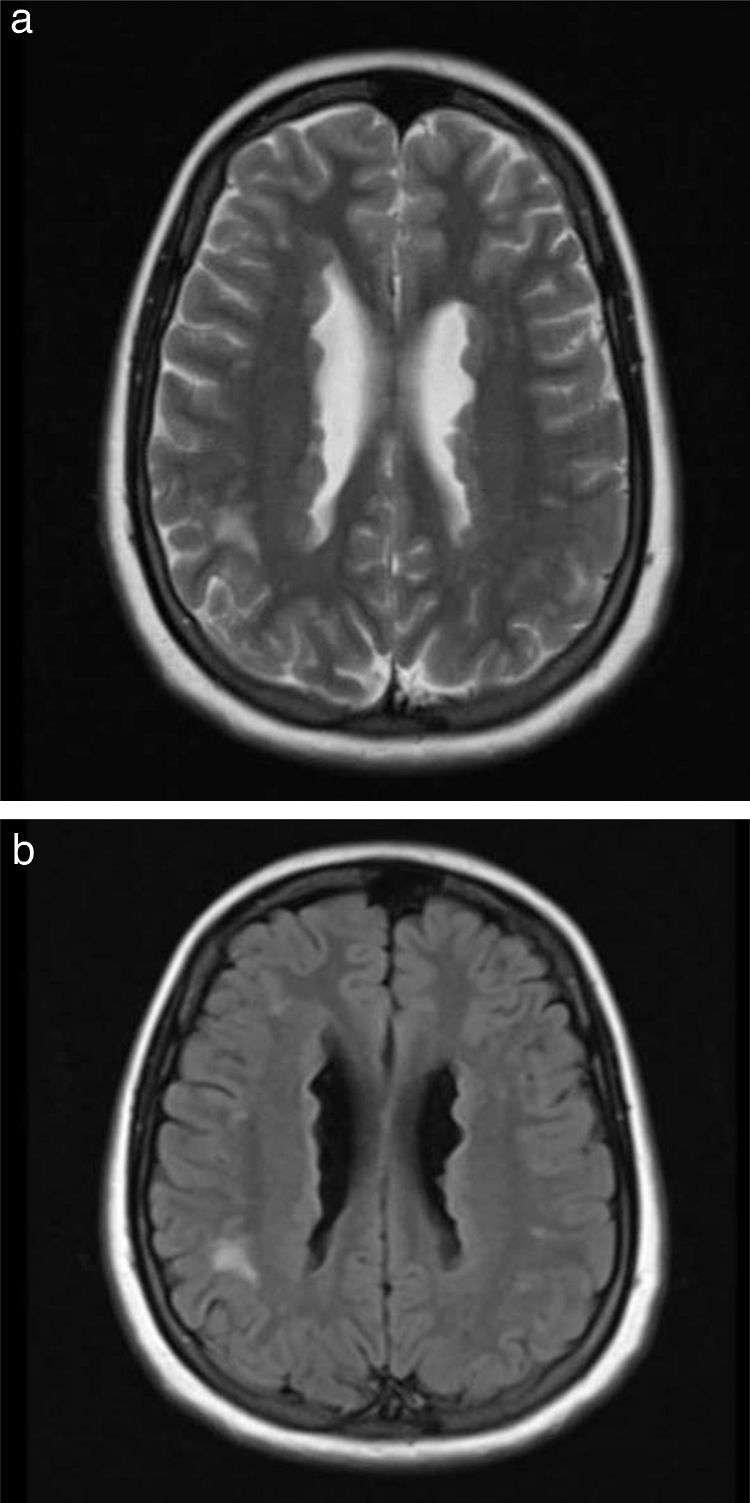
Our patient was taken to the emergency department due to exacerbation of seizures; electroencephalography and metabolic, toxicity, and microbiology studies yielded normal results. Furthermore, a brain MRI scan revealed an irregularity in the contour of the lateral ventricles, which appeared hyperintense on T2-weighted sequences (Fig. 1), suggestive of PNH. Considering that the patient was in the first trimester of pregnancy, phenobarbital was suspended and she started treatment with oral levetiracetam dosed at 500 mg every 12 h, which partially improved seizure frequency.
 Axial T2-weighted sequence. (b) Axial T2-weighted FLAIR sequence suggesting PNH.")
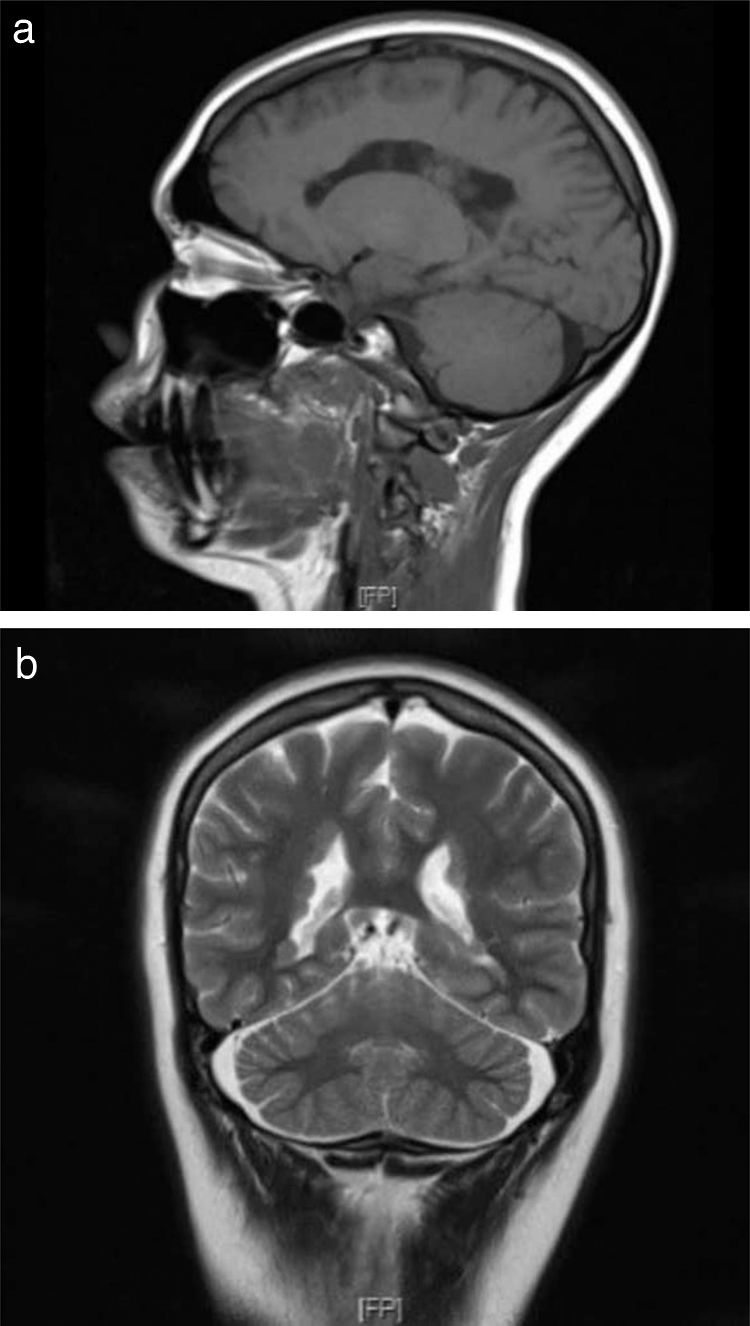
First-degree relatives with history of epilepsy were studied, revealing PNH in the patient’s mother (Fig. 2a). Both patients underwent neuropsychological tests, which yielded normal results.
 Sagittal T1-weighted sequence (patient’s daughter). (b) Coronal T2-weighted sequence showing a hypointense lesion causing irregularities in the external borders of the lateral ventricles due to PNH (patient’s mother).")
The patient’s 9-month-old daughter also underwent a brain MRI study, revealing findings compatible with PNH (Fig. 2b). We recommended a genetic study, and genetic sequencing revealed the hemizygous presence of a probably pathogenic variant (c.7512_7528+8del25) of the FLNA gene; this variant affects the splice donor site, which alters mRNA processing, and is not yet included in the different databases consulted.
None of the patients presented any other type of malformation of the central nervous system or cardiac system.
Genetic testing confirmed diagnosis of this infrequent disease known as familial bilateral PNH, following an X-linked inheritance pattern, caused by a mutation in the FLNA gene, which presented a probably pathogenic variant that has not previously been reported in the literature. This contributes to our understanding of the disease and is useful for genetic counselling.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Angulo-Maldonado M, Lara-Sarabia O, Cadena-Bonfanti A, Ulloa-Piza A. Heterotopia nodular periventricular bilateral hereditaria ligada al cromosoma X. Neurología. 2022;37:232–234.
