



Dear Editor:
Autosomal dominant polycystic kidney disease, caused by mutations in either PKD1 or PKD2, is the most frequent hereditary kidney disease and one of the most causes of end-stage kidney disease.1
Cavernous angiomas or cavernous cerebral malformations represent up to 13% of cerebral vascular lesions and consist of abnormally enlarged capillary cavities surrounded by a single layer of endothelium and devoid of brain parenchyma.2,3 With a prevalence of 0.1%–0.5%, they can be single or multiple and sporadic or familial.2,3 Familial multiple cavernomatosis accounts for up to 50% and usually presents with multiple lesions in 84% compared to 15%–25% of sporadic cases.2,3 Development of cavernous cerebral malformations is linked to mutations within KRIT1 (CCM1), CCM2, and PDCD10 (CCM3). Cavernous cerebral malformations can manifest as cerebral hemorrhage, headache, seizures, and progressive or transient focal neurologic deficits.4
Only 2 cases with autosomal dominant polycystic kidney disease and cavernous cerebral malformations have been reported.4,5 Herein, we report the third case of cavernous cerebral malformation associated with autosomal dominant polycystic kidney disease and cerebellopontine angle syndrome.
A 27-year-old Indian man presented to the outpatient department with sudden onset dizziness, gait unsteadiness, and a sense of “buzzing” sounds inside his right ear, followed by malalignment of eyes (squint), deviation of angle of mouth to the left, drooling of saliva from her right angle of the mouth, reduced taste sensations and salivation, and incomplete closure of her right eyelids for the last 2 days. The next day, he noticed a sudden onset of decreased sensation over the left half of the body and right half of the face following an episode of vomiting.
Neurological examinations revealed loss of all sensations over the forehead and face of the right side, paralysis of right-sided muscles of mastication, paralysis of right lateral rectus muscle producing internal strabismus and horizontal diplopia (especially when looking towards the right side), right-sided lower-motor-neuron type facial paralysis, right-sided sensorineural hearing loss, and reduced sensations over the left half of the body. Left-sided deep tendon reflexes were brisk, and the left-sided plantar response was extensor. The remaining neurological examination was normal.
His past medical history revealed he had received treatment for 4 episodes of severe abdominal pain with the passage of cola-colored urine that indigenous medical practitioners treated as “urinary tract infections”. Although most of his family members had never suffered from any neuropsychiatric ailments except his elder brother, who was diagnosed to be having a Brown-Sequard syndrome-like presentation following a hemorrhagic transformation of spinal cord cavernous malformation, and who later died of chronic renal failure at the age of 36 years. His father had also died at the age of 40 years with a diagnosis of chronic renal failure.
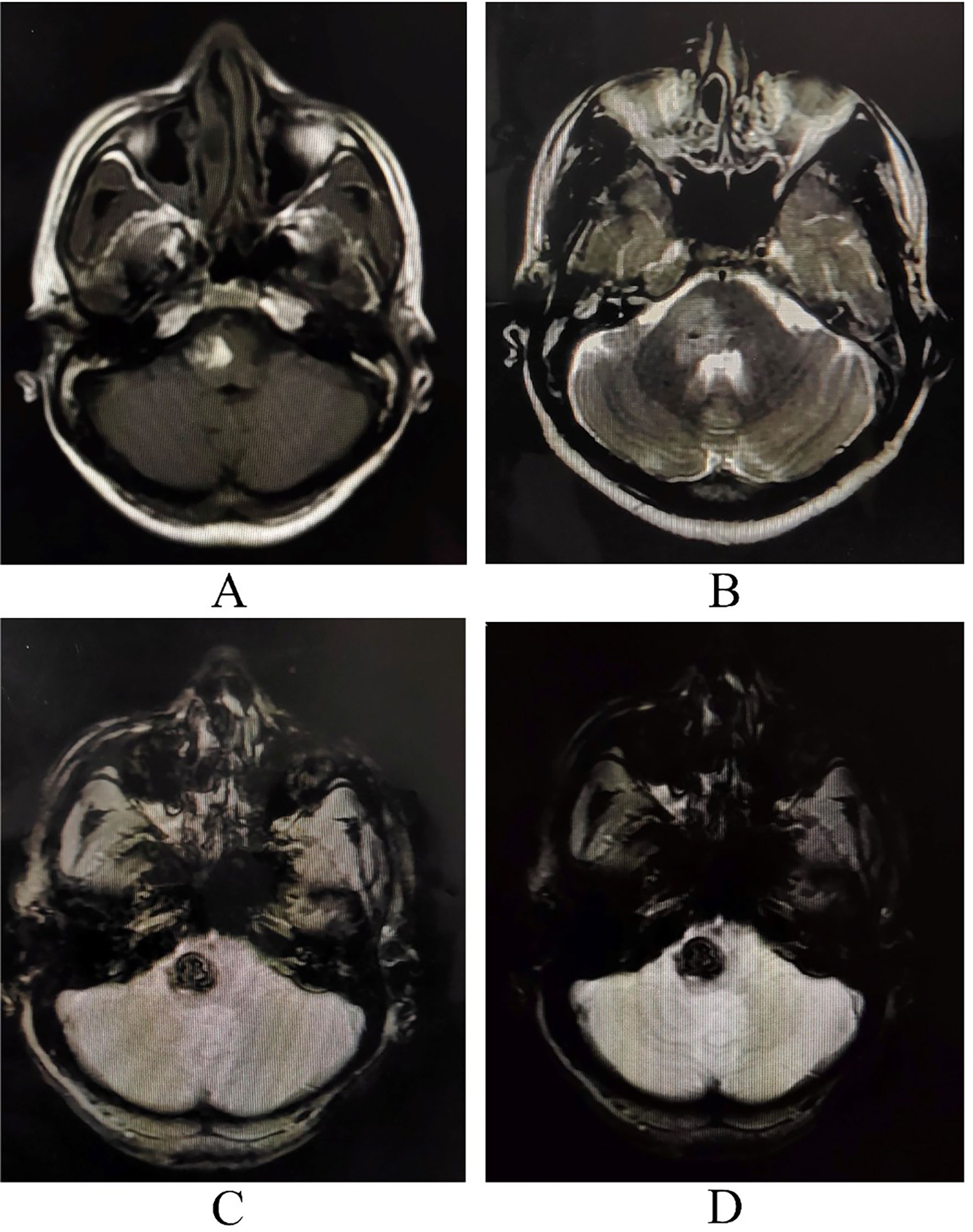
A cerebellopontine angle lesion was suspected and neuroimaging was ordered. Magnetic resonance imaging (MRI) of the brain revealed a right side lesion of pons suggestive of cavernous cerebral malformation (Fig. 1). MRI of the spinal cord revealed no abnormalities. Cerebral digital subtraction angiography ruled out any arteriovenous malformation. Complete metabolic panel and other relevant investigations were within normal limits. Ultrasonography of the kidneys revealed that he had polycystic kidney disease. Two of his sisters and paternal uncles were screened, and they were found to harbor polycystic kidney disease. Besides, when these same family members were screened for the presence of cavernous, one of the paternal uncles and one sister had solitary asymptomatic cavernous cerebral malformations.
 and T2-weighted imaging (B) with signal blooming on GRE/SWI (C and D) sequences on the right side of pons with surrounding well-circumscribed edema extending up to upper medulla and right middle cerebellar peduncles and mild mass effect over the fourth ventricle, suggestive of cavernous cerebral malformation with recent hemorrhage within the lesion.")
Magnetic resonance imaging of the brain revealing altered intensity lesion hyper on T1- (A) and T2-weighted imaging (B) with signal blooming on GRE/SWI (C and D) sequences on the right side of pons with surrounding well-circumscribed edema extending up to upper medulla and right middle cerebellar peduncles and mild mass effect over the fourth ventricle, suggestive of cavernous cerebral malformation with recent hemorrhage within the lesion.
Because of the favorable clinical evolution and reversal of neurological symptoms and signs, he was discharged after 15 days of hospitalization, with continued vigilance and indication for a repeated MRI. Albeit excision of the cavernous angioma was proposed, it could not be performed due to our institute's lack of advanced neurosurgical facilities. Mutational analysis was planned, but it could not be accomplished again due to financial restraints.
MRI, especially gradient-echo sequences, is the most sensitive test for cavernous cerebral malformations detection. The characteristic image is “popcorn”, with a well-defined reticulated nucleus, heterogeneous signal due to blood in different stages, and a hypointense rim of hemosiderin.3 Most cavernous cerebral malformations are supratentorial, although they can also be found in the brainstem and spinal cord.3
As far as we know, this is the first Indian case of probable familial solitary cavernous cerebral malformation associated with autosomal dominant polycystic kidney disease presenting with cerebellopontine angle syndrome as a new clinical and radiological manifestation.
The hypothesis of the 2-hit mechanism or “in 2 steps” has gained importance in recent studies on cavernous cerebral malformations pathogenesis: the loss or mutation of the 2 alleles of the gene would be necessary to suffer from the disease.1–3 New data derived from clinical research using molecular genetics and advanced imaging techniques have enhanced tools for assessing the diagnosis and prognosis of patients with autosomal dominant polycystic kidney disease and cavernous cerebral malformations and their families.1,6
It is essential to carry out the genetic study and radiological follow-up of possible asymptomatic carriers with relatives affected by familial or sporadic multiple cavernomatosis to study new genetic alterations, clinical follow-up, and early treatment since it improves neurological symptoms. It contributes to a better prognosis of this potentially life-threatening treatable illness. Nevertheless, the major limitation of this study is not having performed the genetic confirmation because of limited financial resources.
Study fundingNil.
DisclosuresDr. Ritwik Ghosh (ritwikmed2014@gmail.com) reports no relevant disclosures.
Dr. Moisés León-Ruiz (pistolpete271285@hotmail.com) reports no relevant disclosures.
Dr. Dipayan Roy (dipayan.1993@yahoo.in) reports no relevant disclosures.
Dr. Julián Benito-León (jbenitol67@gmail.com) reports no relevant disclosures.
Ethics statementWritten informed consent was obtained from the patient to publish this case report and any accompanying images.
Author contributionsAll authors contributed significantly to the creation of this manuscript; each fulfilled criterion as established by the ICMJE.
J. Benito-León is supported by the National Institutes of Health, Bethesda, MD, USA (NINDS #R01 NS39422), the European Commission (grant ICT-2011-287739, NeuroTREMOR), the Ministry of Economy and Competitiveness (grant RTC-2015-3967-1, NetMD—platform for the tracking of movement disorder), and the Spanish Health Research Agency (grant FIS PI12/01602 and grant FIS PI16/00451).
