





Las crioglobulinas son inmunoglobulinas que se pueden encontrar en el plasma, aisladas o formando complejos inmunitarios, y precipitan a temperaturas inferiores a los 37°C. Ciertas infecciones y enfermedades reumáticas o hemáticas pueden acompañarse de la presencia de estas proteínas en el suero, lo que da lugar a las crioglobulinemias. En ocasiones, estas pasan inadvertidas por completo por ser asintomáticas, pero otras veces pueden aparecer determinadas manifestaciones clínicas en diversos órganos y sistemas, entre ellos la piel.
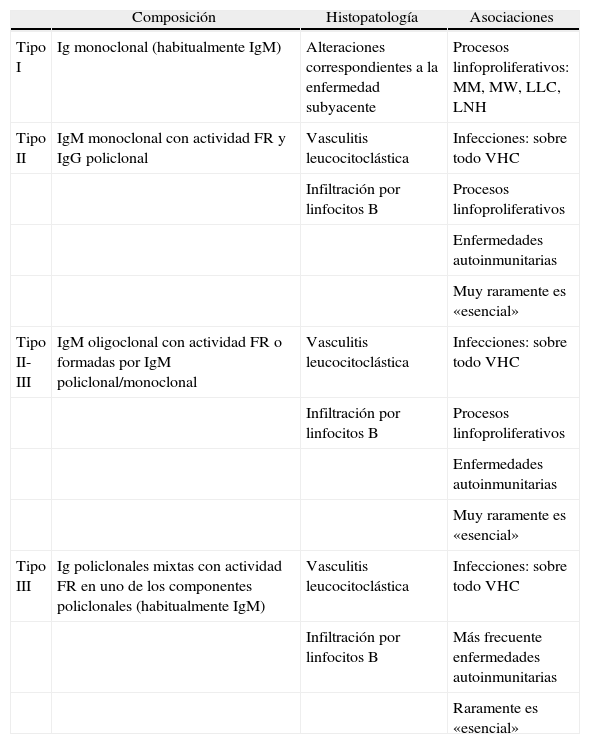
ClasificaciónLas crioglobulinemias se clasifican clásicamente según la composición de sus crioglobulinas en tres subtipos: I, II y III. En la crioglobulinemia de tipo I, las crioglobulinas se encuentran formadas por un solo subtipo de inmunoglobulina, con más frecuencia IgM, aunque también IgG, IgA o incluso cadenas ligeras. En las crioglobulinemias tipos II y III o crioglobulinemias mixtas (CM), las crioglobulinas son complejos inmunitarios formados por IgG policlonal e IgM monoclonal (tipo II) o policlonal (tipo III) respectivamente. La IgM se comporta como autoanticuerpo y exhibe actividad de factor reumatoide (FR). Con técnicas de laboratorio más sensibles se han detectado otros tipos de crioglobulinas además de las anteriores (tipo II-III). Estas aparecen en formas intermedias de CM o en formas evolutivas o de transformación entre una crioglobulinemia tipo III y una tipo II (tabla 1)1.
Clasificación y características clinicopatológicas de las crioglobulinemias.
| Composición | Histopatología | Asociaciones | |
| Tipo I | Ig monoclonal (habitualmente IgM) | Alteraciones correspondientes a la enfermedad subyacente | Procesos linfoproliferativos: MM, MW, LLC, LNH |
| Tipo II | IgM monoclonal con actividad FR y IgG policlonal | Vasculitis leucocitoclástica | Infecciones: sobre todo VHC |
| Infiltración por linfocitos B | Procesos linfoproliferativos | ||
| Enfermedades autoinmunitarias | |||
| Muy raramente es «esencial» | |||
| Tipo II-III | IgM oligoclonal con actividad FR o formadas por IgM policlonal/monoclonal | Vasculitis leucocitoclástica | Infecciones: sobre todo VHC |
| Infiltración por linfocitos B | Procesos linfoproliferativos | ||
| Enfermedades autoinmunitarias | |||
| Muy raramente es «esencial» | |||
| Tipo III | Ig policlonales mixtas con actividad FR en uno de los componentes policlonales (habitualmente IgM) | Vasculitis leucocitoclástica | Infecciones: sobre todo VHC |
| Infiltración por linfocitos B | Más frecuente enfermedades autoinmunitarias | ||
| Raramente es «esencial» |
FR: factor reumatoide; LLC: leucemia linfática crónica; LNH: linfoma no hodgkiniano; MM: mieloma múltiple; MW: macroglobulinemia de Waldeström; VHC: virus hepatitis C. Modificado de: Ferri C. Mixed cryoglobulinemia. Orphanet J Rare Dis. 2008;3:25.
La prevalencia global de las crioglobulinemias no ha sido establecida hasta el momento. En cuanto a la etiología, hay una variedad de trastornos inmunitarios, hemáticos, neoplásicos e infecciosos que pueden asociarse a su aparición. Con mucha diferencia, la crioglobulinemia más frecuente es la tipo II, que representa un 50-60% de los casos2 y en el pasado se conocía como crioglobulinemia esencial. Su relación con la infección por el VHC quedó manifiesta con el descubrimiento del virus a principios de los años noventa3,4 y se calcula que en el área mediterránea el 90% de los pacientes con CM se encuentran infectados por dicho agente infeccioso. El VHC, por lo tanto, se relaciona habitualmente con la crioglobulinemia tipo II y, menos frecuentemente, con la III. No obstante, existen otras causas de CM, como ciertas infecciones (VIH, VHB, etc.) o enfermedades autoinmunitarias (LES, síndrome de Sjögren, esclerosis sistémica)5. La crioglobulinemia tipo I corresponde a un 10-15% de los pacientes con crioglobulinemia y puede encontrarse en el contexto de determinados trastornos linfoproliferativos, como la macroglobulinemia de Waldeström, el mieloma múltiple o la leucemia linfática crónica2.
Crioglobulinemia mixta y VHCLa infección por el VHC afecta a mecanismos inmunológicos fundamentales y predispone a la aparición de trastornos autoinmunitarios y linfoproliferativos. Entre ellos, el principal y más frecuente es la crioglobulinemia tipo II, pero existen otros como la glomerulonefritis membranoproliferativa, el síndrome seco, la artritis reumatoide o algunos linfomas no hodgkinianos, por citar sólo algunos2.
La incidencia estimada de CM en pacientes infectados por el VHC varía enormemente en función de las series entre el 10 y el 70%5. No obstante, parece ser que es más frecuente en los pacientes con mayor tiempo de evolución de la infección. La CM es mucho más prevalente en el sur de Europa que en el norte de Europa o en Estados Unidos, de manera que el 60% de los pacientes europeos infectados por el VHC tienen crioglobulinemia mixta, mientras que la prevalencia es bastante menor en Estados Unidos6,7. Algunos estudios realizados en Italia, Francia y China demostraron una asociación con ciertos alelos del HLA como el DRB1*11, DR3, DR5 y DR68, pero en estudios realizados en Japón no se pudo demostrar ninguna asociación significativa entre el HLA y la existencia de CM9.
Etipatogenia; el papel del VHCComo se ha comentado, la infección crónica por el VHC conduce a alteraciones cruciales en determinados mecanismos inmunológicos, con la consecuente acumulación de complejos inmunitarios circulantes y la aparición de fenómenos autoinmunitarios. El hecho fundamental en la CM parece ser la expansión clonal, estimulada directamente por el virus, de linfocitos B con expresión de FR en el hígado, ganglios linfáticos y sangre periférica, de manera que se la considera un trastorno linfoproliferativo10. Lo que no está claro es por qué mecanismos los linfocitos B inician su desregulación en el contexto de la infección. Sea como fuere, la producción de IgM con actividad FR por esos linfocitos conduciría a la formación de complejos inmunitarios crioprecipitables formados por esta inmunoglobulina, el core del virus e IgG específicas anti-core. El enorme complejo inmunitario resultante tendría la capacidad de unirse de forma específica a las células endoteliales a través del receptor C1q, con la consecuente activación de la cascada del complemento y la producción de una vasculitis11.
ClínicaLas manifestaciones clínicas de las crioglobulinemias son muy heterogéneas y variadas. En el caso de la crioglobulinemia tipo I, que es asintomática en muchas ocasiones12, lo más frecuente es que aparezca acrocianosis, fenómeno de Raynaud o gangrena secundarios a alteraciones en la microcirculación2.
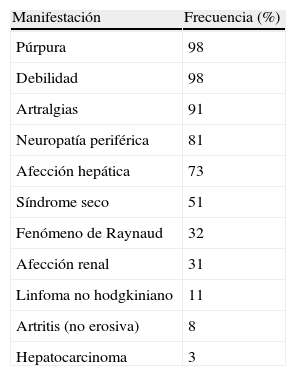
La tríada clínica clásica de la CM es la presencia de púrpura, artralgias y astenia13, y el sustrato histopatológico es el de una vasculitis leucocitoclástica de vasos de pequeño-mediano calibre. No obstante, aunque típica, esta tríada es rara y muchos de los pacientes con CM se encuentran asintomáticos o se presentan sólo con púrpura palpable, en la mayoría de los casos transitoria. En este sentido, estas crioglobulinemias se clasifican en el grupo de las vasculitis de pequeño vaso, junto con la vasculitis leucocitoclástica o la púrpura de Schönlein-Henoch. De este modo, bajo el sustrato de una vasculitis, pueden aparecer múltiples manifestaciones clínicas secundarias a la afección de otros órganos diana, como riñones, sistema nervioso periférico o tracto gastrointestinal, y también pueden aparecer linfadenopatías generalizadas14. Por otro lado, en caso de que haya gran cantidad de crioglobulinas circulantes, pueden aparecer manifestaciones clínicas secundarias a un síndrome de hiperviscosidad. Además, en el contexto de una CM, es posible el desarrollo de neoplasias como el hepatocarcinoma o el carcinoma papilar de tiroides12 y de procesos linfoproliferativos secundarios a la expansión y estimulación crónica de los linfocitos B15. A continuación se revisan las manifestaciones clínicas de la CM (tabla 2).
Manifestaciones clínicas de la crioglobulinemia mixta.
| Manifestación | Frecuencia (%) |
| Púrpura | 98 |
| Debilidad | 98 |
| Artralgias | 91 |
| Neuropatía periférica | 81 |
| Afección hepática | 73 |
| Síndrome seco | 51 |
| Fenómeno de Raynaud | 32 |
| Afección renal | 31 |
| Linfoma no hodgkiniano | 11 |
| Artritis (no erosiva) | 8 |
| Hepatocarcinoma | 3 |
Modificado de: Ferri C. Mixed cryoglobulinemia. Orphanet J Rare Dis. 2008;3:25.
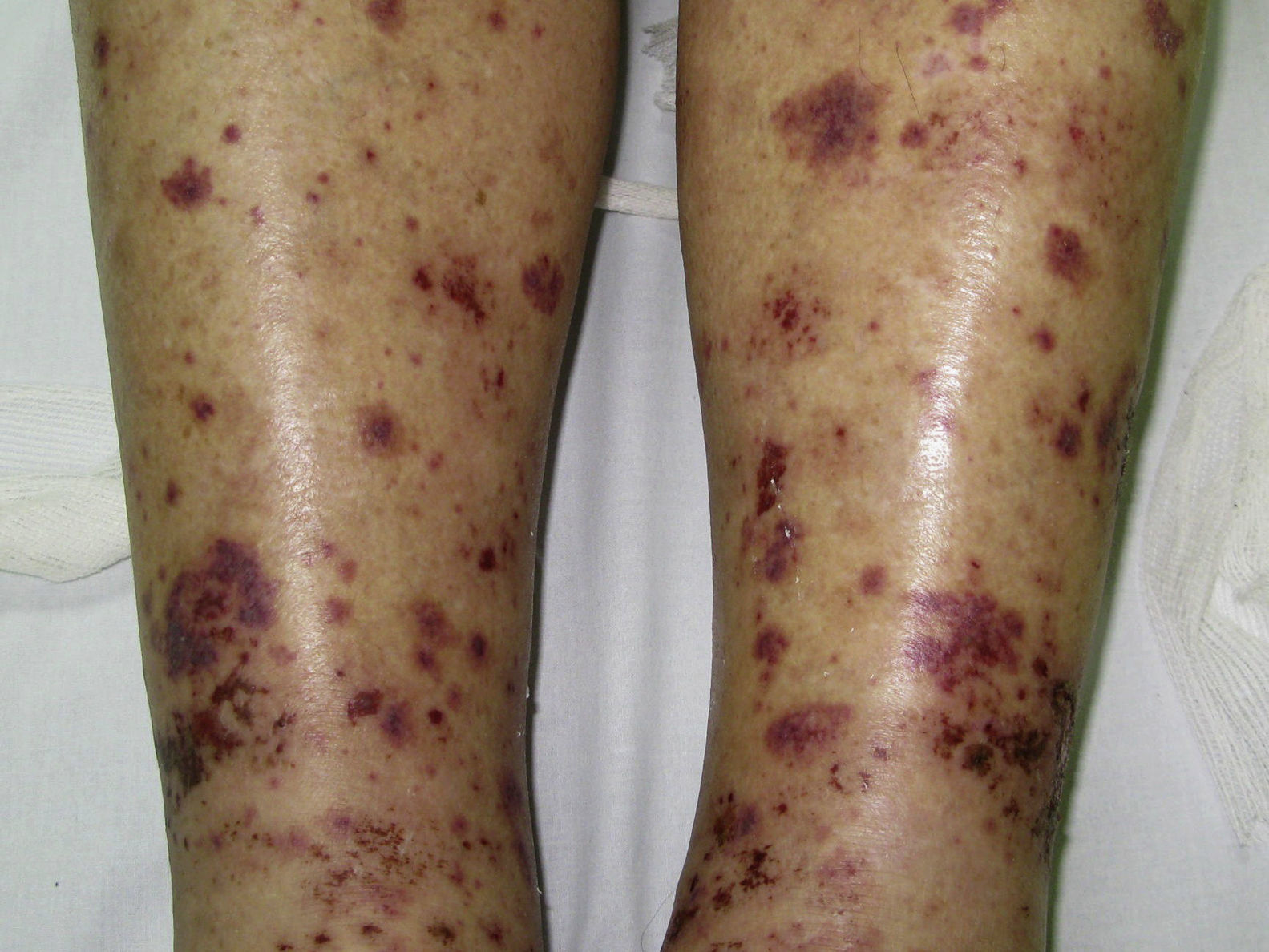
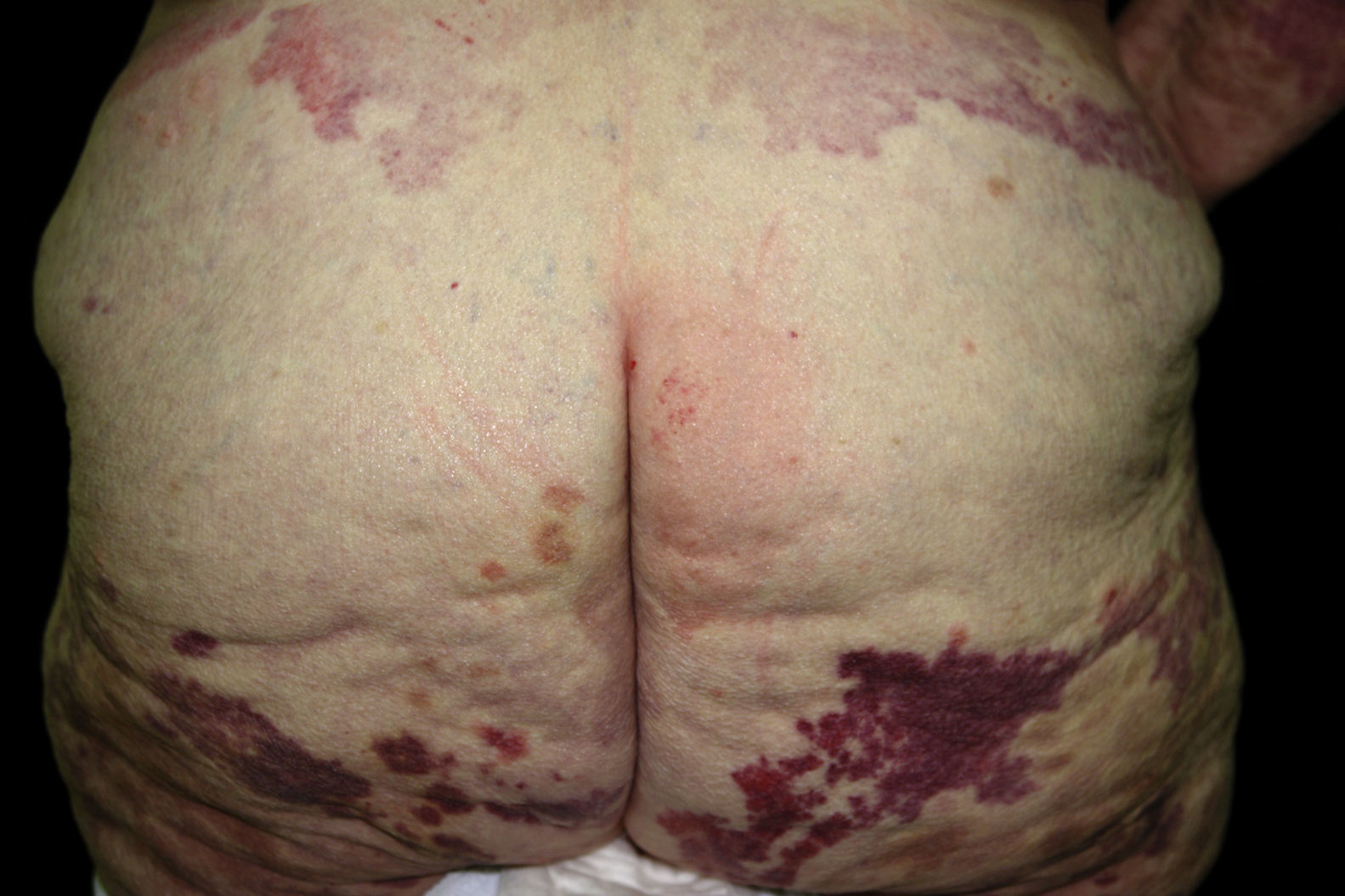
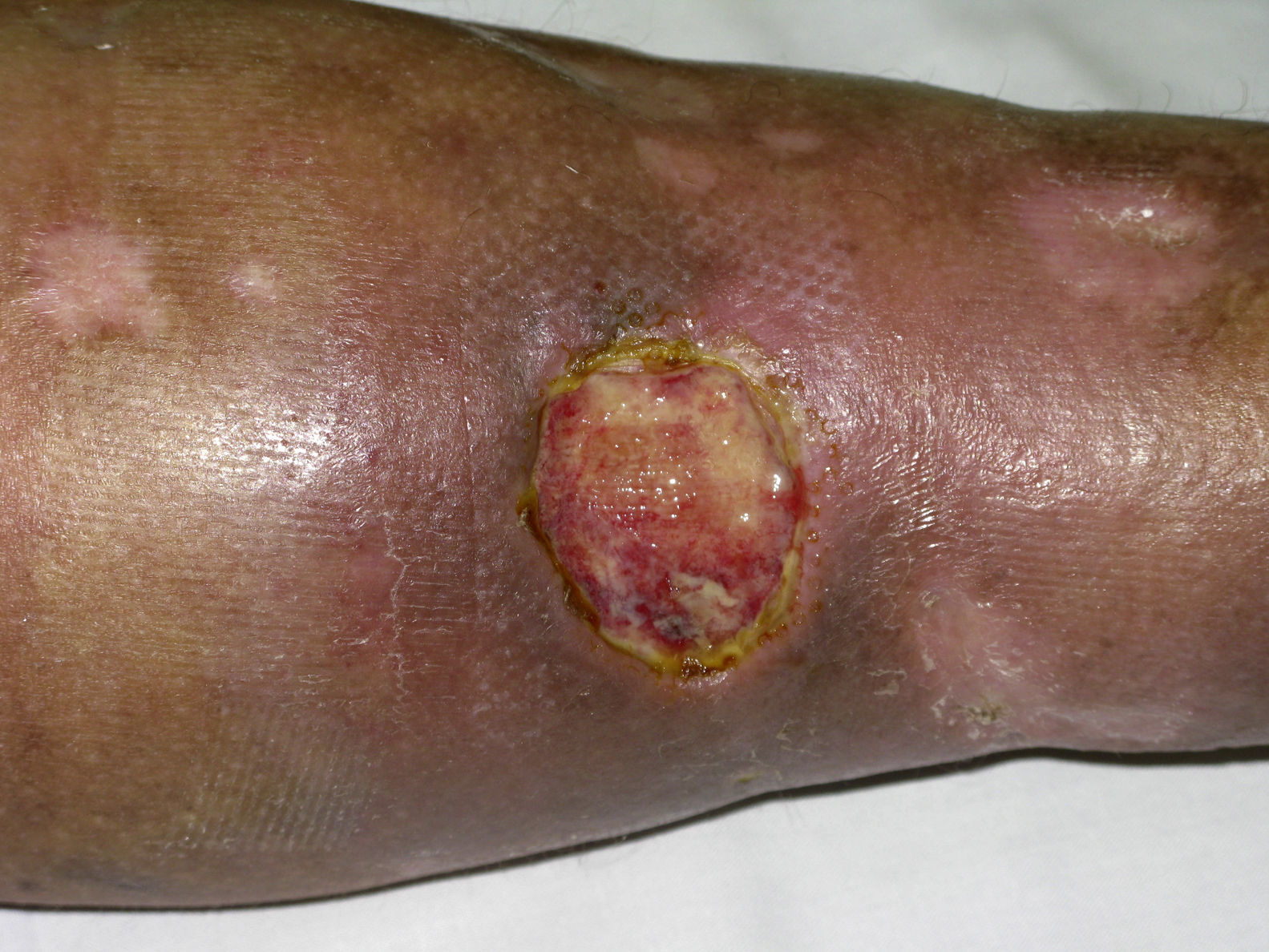
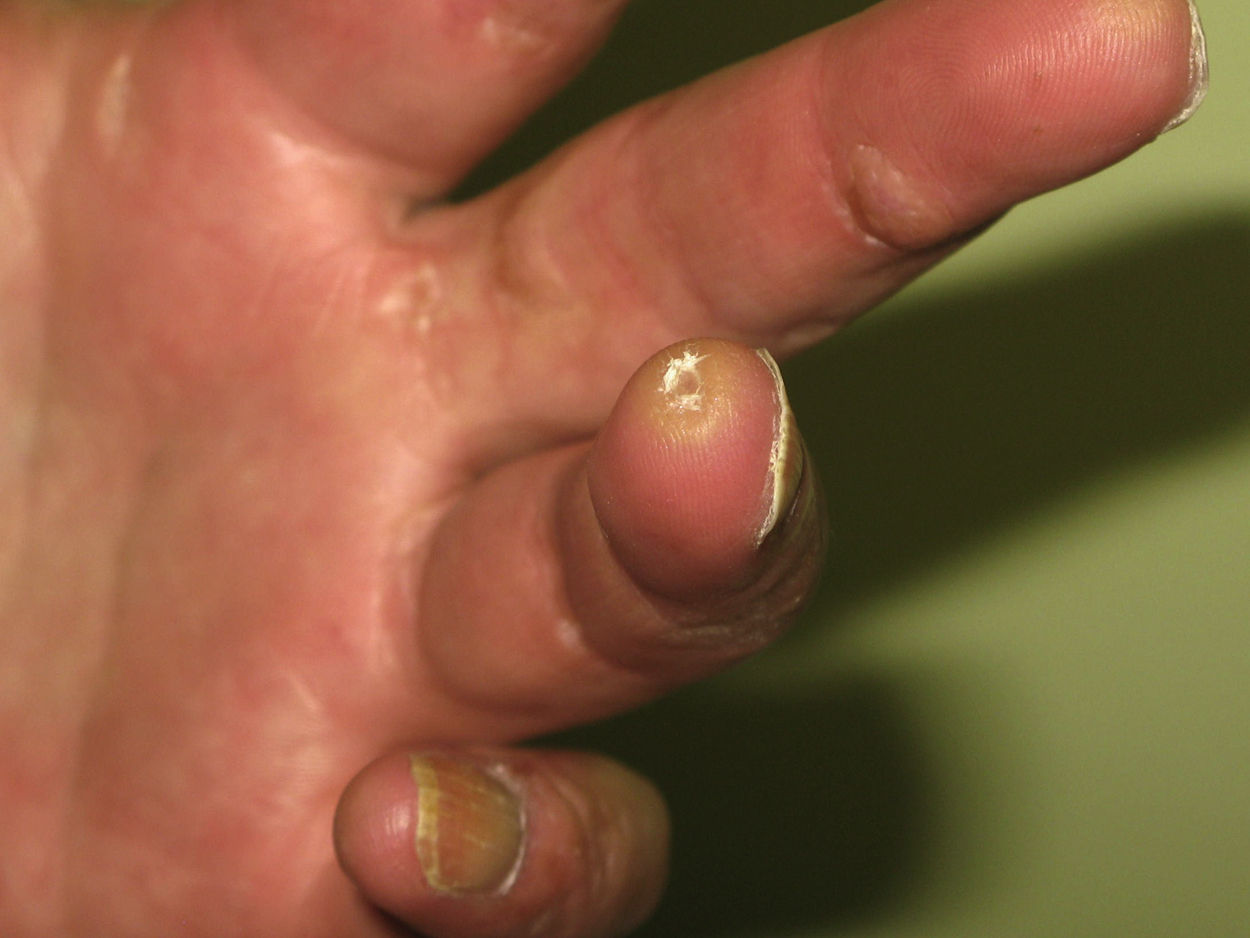
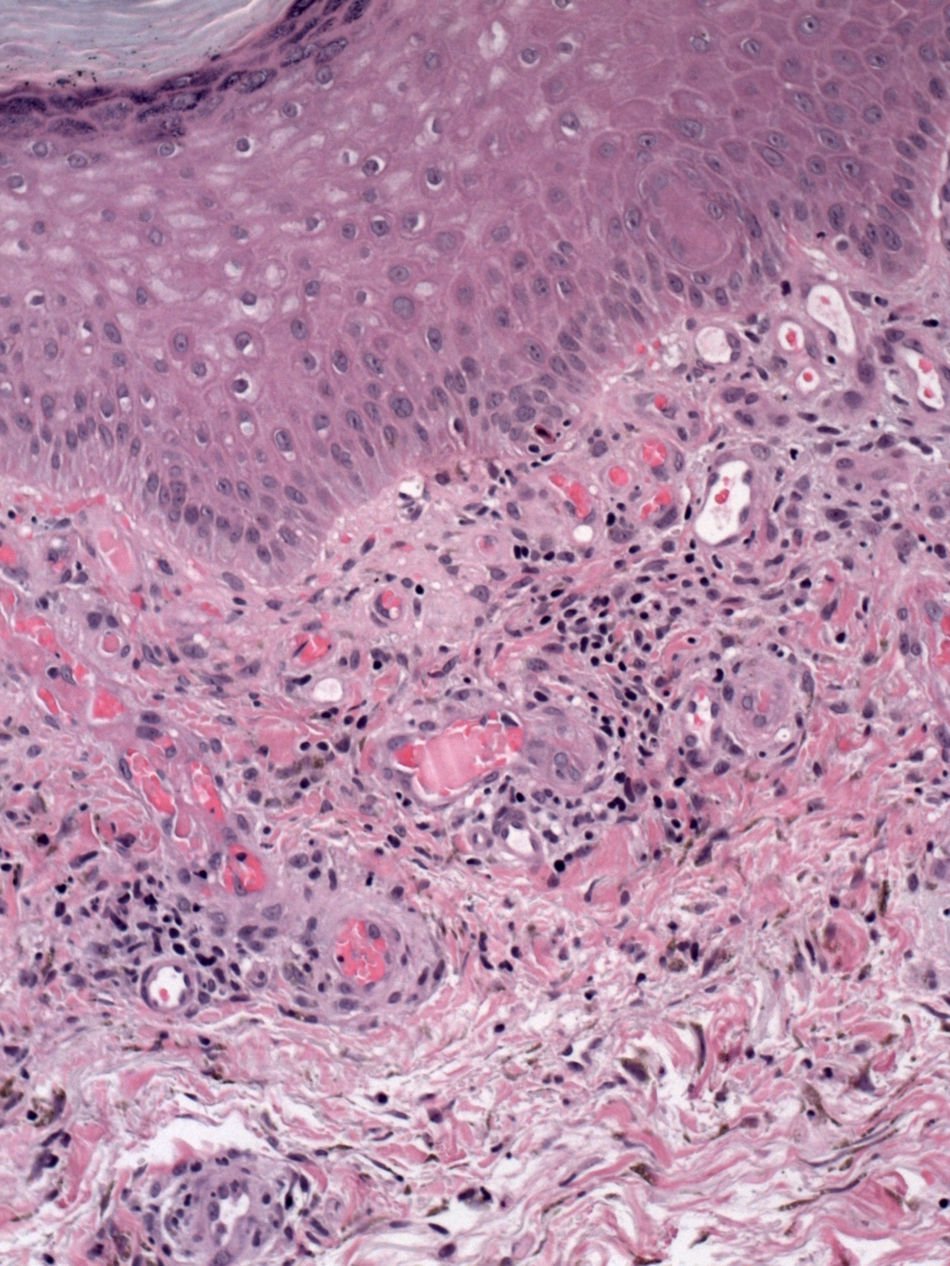
Las lesiones cutáneas son la manifestación clínica más frecuente de la CM12, de manera que las formas leves de la enfermedad suelen cursar con afección cutánea en ausencia de enfermedad sistémica. En la piel, las tres lesiones más típicas son la púrpura palpable (fig. 1), la pigmentación reticulada y la vasculitis urticarial (fig. 2). Con mucho, la púrpura palpable intermitente es la lesión que se observa con más frecuencia (el 90% de los casos sintomáticos)2, de intensidad y extensión muy variables, y que suele representar la primera manifestación de la enfermedad. Por lo general, las lesiones se encuentran localizadas en la región distal de las extremidades inferiores y suelen aparecer en las últimas horas del día coincidiendo con la bipedestación o sedestación prolongadas16. Como consecuencia de los brotes repetidos, y como ya se ha señalado, los pacientes con CM acaban presentando áreas persistentes de dermatitis ocre o pigmentación reticulada. Otras lesiones que pueden observarse en los individuos afectos son úlceras de evolución tórpida localizadas en las piernas y zonas maleolares (fig. 3), púrpura retiforme, fenómeno de Raynaud, urticaria a frigore17, acrocianosis y úlceras isquémicas o gangrena en las zonas acras de los dedos14 (fig. 4). Existen otras formas más graves de la enfermedad, con afección vasculítica difusa de la piel y de dos o más órganos internos, también por la propia vasculitis, pero son mucho más raras. En estos casos las lesiones en la piel denotan también mayor gravedad y suelen consistir en úlceras hemorrágicas y necrosis2.




El sustrato histopatológico es el de una vasculitis leucocitoclástica de vasos de pequeño o mediano calibre y, en ocasiones, se pueden encontrar crioprecipitados intramurales PAS positivos (fig. 5). Mediante inmunofluorescencia directa, es posible observar depósitos de IgM-FR, IgG y C3 en la pared de los vasos afectos14.
Manifestaciones reumáticas
Los pacientes con crioglobulinemia mixta suelen presentar artralgias y en ocasiones mialgias, pero es relativamente rara la existencia de una artritis franca2. Sobre todo afecta a las manos, las rodillas y los codos y puede asociarse a la exposición al frío14. Asimismo, la mitad de ellos aquejan xerostomía y xeroftalmia, aunque sólo un mínimo porcentaje cumple los criterios para el diagnóstico de síndrome de Sjögren12.
Manifestaciones neurológicasCon frecuencia se encuentra una neuropatía periférica en el curso de la CM. En la mayoría de los casos, se presenta en forma de una neuropatía sensitiva de intensidad leve-moderada, que cursa con parestesias, dolor o sensación de quemazón, localizada con frecuencia en las extremidades inferiores y que se suele exacerbar por la noche12. La incidencia de este tipo de neuropatía, afectando a las piernas y en forma de dolor, parestesias y debilidad muscular, alcanza al 60% de los casos2. El curso suele ser insidioso y progresivo, y los síntomas son rebeldes a la mayoría de los tratamientos ensayados18. El sustrato histopatológico parece ser una vasculitis de los vasa nervorum, pero también parece que hay daño directo del nervio, mediado por mecanismos inmunitarios2.
Existen otras manifestaciones neurológicas que se han asociado a la enfermedad, aunque con frecuencia mucho menor. Entre ellas destacan los trastornos sensitivomotores como traducción de una mononeuritis asimétrica y también la afección del sistema nervioso central, que parece haberse demostrado, dada la alta frecuencia de alteraciones observadas mediante RM19.
Manifestaciones renalesLa enfermedad renal afecta aproximadamente al 30% de los pacientes y es una de las causas más importantes de morbilidad2. La lesión renal condiciona el pronóstico en la CM y aparece en la mayoría de los casos en forma de una glomerulonefritis membranoproliferativa tipo I. Se trata de una forma de glomerulonefritis mediada por inmunocomplejos y caracterizada por una proliferación mensangial endocapilar, vasculitis de arterias renales de pequeño y mediano calibre y el depósito de IgM, IgG y C3 en el glomérulo, entre otros. Desde el punto de vista clínico, aparece en forma de proteinuria, elevación de la creatinina sérica e insuficiencia renal progresiva20,21.
Manifestaciones pulmonaresDe manera anecdótica, se ha observado algún caso de alveolitis subclínica en pacientes con CM al realizar lavados broncoalveolares. Esto puede predisponer a una mayor susceptibilidad a infecciones bronquiales y, mucho más raramente, a una fibrosis pulmonar intersticial22.
Trastornos endocrinosHay una serie de trastornos endocrinos que parecen ser más frecuentes en los pacientes con CM que en la población general. Entre ellos se encuentran la tiroiditis autoinmunitaria, el hipotiroidismo subclínico y las neoplasias de tiroides12.
Linfoma BSe trata de la neoplasia que con más frecuencia puede complicar el curso de la CM, por lo general en estadios avanzados12. Al parecer el desarrollo de estos trastornos linfoproliferativos estaría en relación con la expansión de linfocitos B y los infiltrados presentes en el hígado o la médula ósea de los pacientes con esta enfermedad. Durante bastante tiempo, a diferencia de los linfomas francos, estas proliferaciones linfoides pueden permanecer sin modificación alguna, de manera que se propuso el término trastornos linfoproliferativos monotípicos de significado incierto (monotypic lymphoproliferative disorder of undetermined significance [MLDUS]) para referirse a ellas. No obstante, durante el curso de una CM, puede desarrollarse un verdadero linfoma de células B, que debe sospecharse con la desaparición de las crioglobulinas y el FR en suero y la existencia de valores anormalmente altos de C415,23. Asimismo, en un estudio publicado recientemente, se ha visto que niveles bajos de gammaglobulina se asociarían también con la existencia de un linfoma24.
Otras neoplasiasLas dos neoplasias sólidas que con más frecuencia pueden desarrollarse en estos pacientes son el hepatocarcinoma y el carcinoma papilar de tiroides12. En este sentido, la CM debe considerarse un síndrome paraneoplásico, de manera que los pacientes deben ser monitorizados adecuadamente para poder establecer el diagnóstico de estas potenciales complicaciones lo antes posible25.
DiagnósticoEn ausencia de unos criterios diagnósticos aceptados universalmente, el hallazgo de crioglobulinas mixtas circulantes y valores bajos de C4 en el suero de un paciente con púrpura debe hacer sospechar la existencia de este síndrome. La demostración de una vasculitis leucocitoclástica de vasos de pequeño o mediano calibre en una biopsia cutánea de una lesión reciente constituye el sustrato histopatológico típico de la enfermedad. En la tabla 3 se recogen los criterios de clasificación de la crioglobulinemia mixta propuestos por el grupo italiano para el estudio de la crioglobulinemia12.
Criterios de clasificación de los pacientes con crioglobulinemia mixta.
| Criterio | Serológico | Patológico | Clínico |
| Mayores | Crioglobulinas mixtas; cifras de C4 bajas | Vasculitis leucocitoclástica | Púrpura |
| Menores | Factor reumatoide (+); VHC (+) o VHB (+) | Infiltrados clonales de linfocitos B (hígado o médula ósea) | Hepatitis crónica, glomerulonefritis membranoproliferativa, neuropatía periférica, úlceras cutáneas |
| Diagnóstico definitivo de crioglobulinemia mixta | 1. Crioglobulinas mixtas en suero (± cifras de C4 bajas)+púrpura+vasculitis leucocitoclástica | ||
| 2. Crioglobulinas mixtas en suero (± cifras de C4 bajas)+dos criterios clínicos menores+2 criterios serológicos/patológicos menores | |||
Modificado de: Ferri C. Mixed cryoglobulinemia. Orphanet J Rare Dis. 2008;3:25.
La extracción de sangre para el estudio de las crioglobulinas debe realizarse en unas condiciones determinadas. Por ello, en la mayoría de los hospitales y centros de salud, estas extracciones se realizan en días concretos. La muestra obtenida mediante punción venosa debe precalentarse a unos 37°C. Posteriormente el suero se separa mediante centrifugación y se mantiene en la nevera a una temperatura de 4°C14. A los 7 días, puede realizarse la determinación del criocrito y el estudio y la caracterización de las crioglobulinas12.
Diagnóstico diferencialEn ocasiones el diagnóstico de CM puede ser difícil por el gran polimorfismo clínico y la similitud con otros trastornos inmunitarios que pueden asociarse a la infección por el VHC en ausencia de CM. Este es el caso de otras enfermedades sistémicas con positividad de FR, como la artritis reumatoide (AR) o el síndrome de Sjögren primario (SS)12. Para el diagnóstico diferencial con la AR, cabe recordar, como ya se ha señalado, que los pacientes con CM suelen presentar artralgias y no artritis erosiva. La determinación de los anticuerpos antipéptido citrulinado, marcador de la AR clásica, puede ser de mucha utilidad26. Por otro lado, la mitad de los pacientes con CM aquejan xerostomía y xeroftalmia, pero la mayoría de ellos no cumplen los criterios para el diagnóstico de SS primario. Por ejemplo, es rara la presencia de anticuerpos anti-Ro/La en la CM, así como los hallazgos histopatológicos típicos en las biopsias de la glándula salival. Además, recientemente se propuso que la presencia de la infección por el VHC se debía considerar criterio de exclusión para el diagnóstico de SS primario27.
PronósticoEl curso de la CM es impredecible y está muy influido por la existencia de enfermedades subyacentes, las complicaciones que pueden ir apareciendo y la respuesta a los diversos tratamientos. El pronóstico es peor en los pacientes con afección renal, procesos linfoproliferativos, neoplasias o insuficiencia hepática, y se estima que la supervivencia media es de aproximadamente un 50-60% a los 10 años tras el diagnóstico. A todos los pacientes afectos de CM, debe efectuarse un seguimiento estricto y monitorización de las potenciales complicaciones vitales16.
TratamientoEl abordaje terapéutico de los pacientes con CM es complejo y debe ser individualizado. En los casos de CM asociada a la infección por el VHC, el tratamiento de elección debe ser aquel que persiga la eliminación del virus y habitualmente se realiza con la combinación de interferón alfa y ribavirina. En los casos en que haya complicaciones graves que contraindiquen este tratamiento (p. ej., una neuropatía periférica grave), se pueden utilizar corticoides sistémicos, plasmaféresis, dietas bajas en antígenos o agentes inmunosupresores. En la CM «esencial», la ciclofosfamida y el rituximab siguen siendo los fármacos de primera línea.
En la práctica diaria, la gravedad de la clínica también determina directamente el abordaje terapéutico que se ha de llevar a cabo. Así, los pacientes con manifestaciones vasculíticas muy intensas deben ser tratados precozmente con altas dosis de corticoides, plasmaféresis, ciclofosfamida o rituximab. Por el contrario, los pacientes asintomáticos o con una púrpura transitoria no precisan de tratamiento alguno, a pesar de que puedan presentar concentraciones muy elevadas de crioglobulinas en el suero2,5,12.