








La esclerosis sistémica (ES) es una enfermedad multisistémica, caracterizada por una fibrosis cutánea extensa, alteraciones vasculares y autoanticuerpos contra varios antígenos celulares. Presenta una importante morbilidad y las tasas de mortalidad más altas entre las enfermedades reumáticas autoinmunitarias, de ahí la importancia de llegar a un diagnóstico adecuado. Contempla gran variabilidad en sus manifestaciones clínicas y, aunque no existen unos criterios diagnósticos bien establecidos, la magnitud o la extensión de la afectación cutánea permiten distinguir las 2 formas clínicas más representativas de la enfermedad: ES cutánea difusa (EScd) y ES cutánea limitada (EScl), que incluso ayudan a establecer un pronóstico. Hasta en el 20% de los pacientes se observan manifestaciones superpuestas con otros procesos autoinmunitarios, lo que da los llamados síndromes de solapamiento u overlap. Por último, son raros, pero existen casos de ES sin clínica cutánea, llamados esclerodermia sin esclerodermia1.
El término esclerodermia hace referencia al endurecimiento de la piel y a la ES. Puede confundirse y aparecer en el contexto de otras enfermedades que se caracterizan por fibrosis cutánea, como la esclerodermia localizada (morfea), el escleredema o la fascitis eosinofílica2. En esta revisión siempre utilizaremos el término que se ocupa como sinónimo de ES.
Hoy por hoy se trata de una enfermedad crónica, aunque se ha avanzado mucho en las últimas décadas, sobre todo en el tratamiento de las complicaciones derivadas de la afectación de órganos internos.
EpidemiologíaLa ES está incluida dentro de las enfermedades raras, con una prevalencia de entre 7 y 489 casos por millón de habitantes y una incidencia de 0,6 a 122 casos por millón de personas y por año. La edad media de aparición es entre los 30 y los 50 años (más temprana en la EScd que en la EScl)3 y la supervivencia media desde el diagnóstico es de unos 12 años4. Se ha observado una mayor prevalencia de casos en Australia o EE. UU. respecto a Japón o Europa, con diferencias también en cuanto a latitudes norte-sur. En determinadas áreas se han registrado casos muy por encima de lo esperado, lo que indicaría una agrupación temporoespacial, aunque no se han llegado a identificar factores determinantes. Los resultados de los estudios epidemiológicos son contradictorios dadas las variaciones metodológicas en la recogida de casos y las diferencias geográficas3,4. La afectación de mujeres respecto a hombres es de 3–4:1 y es más frecuente en la raza negra, de hecho, las mujeres jóvenes de raza negra tienen mayor afectación difusa, mayor incidencia de fenómenos inflamatorios y una menor esperanza de vida5. Es una enfermedad infrecuente en los niños y en éstos se encuentran algunas diferencias respecto a los adultos con ES, así, hay más síndromes de solapamiento, más afectación cardíaca (ésta puede ser causante de muerte) y musculoesquelética; las afecciones renal y pulmonar son más raras6. También se detectan con mayor frecuencia que en los adultos los autoanticuerpos anti-PM-Scl y anti-U1RNP, por el contrario, son raros los anticuerpos anticentrómeros (ACA)7,8.
Etiología y patogeniaLa ES es una enfermedad extremadamente compleja. Intervienen diferentes tipos celulares en diferentes microambientes y mediante múltiples vías y mediadores, y todos interaccionan entre sí. Por tanto, es tal la diversidad clínica presente, que en diferentes pacientes y subtipos de ES predominan unos mecanismos u otros. Aspectos claves de la enfermedad son la inflamación, la vascularización y la formación de tejido conectivo1.
Factores ambientalesLa asociación entre factores de riesgo ocupacionales/ambientales y la ES ha sido ampliamente estudiada. Las exposiciones son muchas veces de larga duración y la inadecuada clasificación del tipo de exposición y otras variables pueden sesgar su estimada asociación con la ES. Los factores ambientales deberían clasificarse en ocupacionales (sílice, disolventes orgánicos), infecciosos (virus, bacterias) y no ocupacionales/no infecciosos (fármacos, pesticidas, siliconas)9.
Tampoco se excluye la posibilidad de que la enfermedad surja o se desencadene por un agente tóxico, dadas las similitudes entre la esclerodermia y algunas enfermedades producidas por sustancias tóxicas, como el síndrome del aceite de colza10 o el síndrome mialgia eosinofilia11.
Factores genéticosLa agrupación familiar de la enfermedad, la alta frecuencia de otras afecciones autoinmunitarias en familiares de pacientes afectados de ES y las diferencias fenotípicas entre razas y grupos étnicos indican que existen factores genéticos implicados en la inducción de la esclerodermia12,13.
Hay ciertas asociaciones inmunogenéticas con determinado complejo mayor de histocompatibilidad o HLA (DRB1*1104, DQB1*0301 o DQB1*0501, entre otros) que pueden predecir el fenotipo clínico final en pacientes con estadios tempranos de síndromes de solapamiento14. También se ha observado una correlación entre diferentes haplotipos del HLA clase ii y determinados subtipos de autoanticuerpos (por ejemplo, ACA y HLA-DQB1*0501, antitopoisomerasa y DPB1*1301 o DQB1*0301, anti-PM-Scl y DRB1*0301). Del mismo modo, se han descrito varios alelos (HLA-DRB1*1501, DRB1*0701, DQA1*0102, DQB1*0602) que podrían tener un efecto protector frente a la ES, dado que se han visto disminuidos en pacientes con ES15. El mismo polimorfismo en el gen PTPN22 que se ha asociado a la artritis reumatoide, al lupus eritematoso sistémico (LES) y a otras enfermedades autoinmunitarias también se asocia a la ES, especialmente en pacientes con anticuerpos ACA y antitopoisomerasa-i15. Se ha indicado que alteraciones de los genes del colágeno dérmico lo hacen especialmente susceptible a la unión con anticuerpos antitopoisomerasa-i16,17. Una reciente publicación indica que el HLA DRB1*0802 y el DQA1*0501 pueden ser predictores de mortalidad en la ES asociados a datos clínicos, analíticos y radiológicos18.
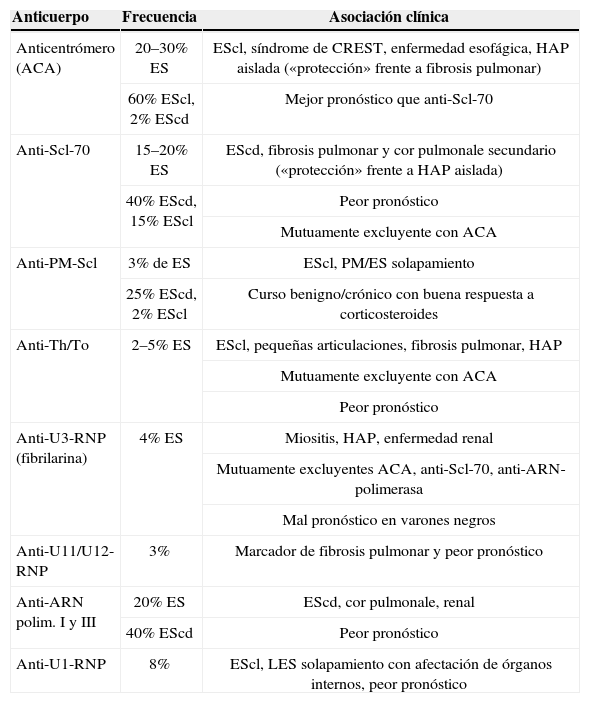
AutoinmunidadEl estímulo inicial para la producción de los autoanticuerpos es desconocido. La mayoría de los pacientes con ES tiene anticuerpos antinucleares (ANA) positivos, que participan en numerosas reacciones características. Cada paciente con ES produce normalmente un solo tipo de autoanticuerpo, lo que sirve de biomarcador para diferentes patrones de afectación cutánea y visceral, al igual que para estimar un pronóstico19 (tabla 1). También se han observado diferentes autoanticuerpos en diferentes grupos étnicos12,13. Aunque está bien establecida su utilidad diagnóstica y pronóstica, no está aclarada su participación en la patogenia de la enfermedad, por lo que este punto es objeto de numerosos estudios.
Autoanticuerpos en la esclerosis sistémica
| Anticuerpo | Frecuencia | Asociación clínica |
| Anticentrómero (ACA) | 20–30% ES | EScl, síndrome de CREST, enfermedad esofágica, HAP aislada («protección» frente a fibrosis pulmonar) |
| 60% EScl, 2% EScd | Mejor pronóstico que anti-Scl-70 | |
| Anti-Scl-70 | 15–20% ES | EScd, fibrosis pulmonar y cor pulmonale secundario («protección» frente a HAP aislada) |
| 40% EScd, 15% EScl | Peor pronóstico | |
| Mutuamente excluyente con ACA | ||
| Anti-PM-Scl | 3% de ES | EScl, PM/ES solapamiento |
| 25% EScd, 2% EScl | Curso benigno/crónico con buena respuesta a corticosteroides | |
| Anti-Th/To | 2–5% ES | EScl, pequeñas articulaciones, fibrosis pulmonar, HAP |
| Mutuamente excluyente con ACA | ||
| Peor pronóstico | ||
| Anti-U3-RNP (fibrilarina) | 4% ES | Miositis, HAP, enfermedad renal |
| Mutuamente excluyentes ACA, anti-Scl-70, anti-ARN-polimerasa | ||
| Mal pronóstico en varones negros | ||
| Anti-U11/U12-RNP | 3% | Marcador de fibrosis pulmonar y peor pronóstico |
| Anti-ARN polim. I y III | 20% ES | EScd, cor pulmonale, renal |
| 40% EScd | Peor pronóstico | |
| Anti-U1-RNP | 8% | EScl, LES solapamiento con afectación de órganos internos, peor pronóstico |
ACA: anticuerpos anticentrómeros; CREST: calcinosis, Raynaud, esofagitis, esclerodactilia y telangiectasias; ES: esclerosis sistémica; EScd: esclerosis sistémica cutánea difusa; EScl: esclerosis sistémica cutánea limitada; HAP: hipertensión arterial pulmonar; LES: lupus eritematoso sistémica; PM: polimiositis.
ANA: presentes en el suero del 75 al 90% de los pacientes con ES, no es específico de ningún subtipo ni confiere matiz pronóstico alguno.
ACA: descritos en 1980, presentes de forma global en el 20–0% de los pacientes con ES, con variaciones raciales. Es raro encontrarlos en gente sana o en otras conectivopatías. Si se detectan en el estudio de un paciente con fenómeno de Raynaud, pueden predecir un futuro desarrollo de ES. También se los ha asociado fuertemente al síndrome de calcinosis, Raynaud, esofagitis, esclerodactilia y telangiectasias (CREST), a la EScl, a la calcinosis y a la pérdida digital isquémica. Los pacientes con ACA tienen menor frecuencia de afectación pulmonar intersticial, pero mayor frecuencia de afectación de hipertensión arterial pulmonar (HAP). Confieren mejor pronóstico que otros autoanticuerpos20.
Antitopoisomerasa-i (originariamente llamada Scl-70): presentes en el 15–20% de los pacientes con ES, no se hallan en personas sanas, con otras conectivopatías o con fenómeno de Raynaud primario. Al igual que con los ACA, la aparición de estos autoanticuerpos en el estudio de un paciente con fenómeno de Raynaud puede predecir el futuro desarrollo de la ES. Están presentes en el 40% de los enfermos con EScd y en menos del 10% con EScl. El 45% de los pacientes con ES y fibrosis pulmonar presentan positividad para estos autoanticuerpos. Confiere mayor riesgo de mortalidad y mayores tasas de fallo cardíaco derecho por fibrosis pulmonar. No está justificado monitorizarlo en el seguimiento del paciente. Los ACA y la antitopoisomerasa-i son mutuamente excluyentes, sólo se detectan simultáneamente en el 0,5% de los pacientes con ES20,21.
Anticuerpos antinucleolares: es un grupo heterogéneo de autoanticuerpos mutuamente excluyentes. Están presentes en el 15–40% de los pacientes con ES. No se observan en sujetos sanos, pero son menos específicos para ES porque se han detectado también en pacientes con LES o síndrome de Sjögren. Anti-PM-Scl: su presencia es variable dependiendo de los grupos estudiados. Aparecen en el 50% de los pacientes con solapamiento polimiositis/ES y sólo en el 2% de enfermos con ES, normalmente la EScl. Confieren un curso más crónico y benigno, con respuesta a dosis bajas/medias de corticoides. Anti-Th/To: presentes en el 2–5% de los pacientes con ES, más frecuentes en Japón y no detectados en personas sanas. También aparecen en enfermos con LES, polimiositis (PM) y fenómeno de Raynaud primario. Suelen asociarse a la EScl, aumentan el riesgo de afectación pulmonar y confieren peor pronóstico. Son mutuamente excluyentes con los ACA. Anti-ARN-polimerasa I, II y III: detectados en el 20% de los pacientes con ES. Los tipos i y iii son más específicos para ES que el tipo ii, y aparecen prácticamente siempre juntos. Están asociados a la afectación cutánea difusa. Se demuestran en el 40% de los pacientes con EScd. Predice mayor riesgo de mortalidad por cor pulmonale y afectación renal. Anti-U3-RNP: presentes en el 4% de los pacientes con ES, mutuamente excluyente con ACA, anti-Scl-70 y anti-ARN-polimerasa. Están asociados a EScd, miositis, hipertensión pulmonar (HTP), enfermedad renal. Anti-U11/12-RNP: aparecen en el 3% de los enfermos con ES, asociados a un mayor riesgo de fibrosis pulmonar, lo que empeora el pronóstico22.
Recientemente se han descrito los anticuerpos anti-PDGF, que parecen tener un efecto estimulador, ya que con su unión al receptor del factor de crecimiento derivado de las plaquetas (PDGF) desencadenan una cascada de señales; esto está aún en estudio23,24.
Los anticuerpos anti-β2-glucoproteína i, especialmente el tipo IgA, se han asociado a un riesgo incrementado de pérdida digital isquémica y a evidencia ecocardiográfica de HAP25.
Anticuerpos anti-IFI16: hay expresión del IFI16 en las células endoteliales y en el epitelio escamoso estratificado, lo que coincide con algunas de las dianas de la ES. Estos autoanticuerpos también parecen afectar al fenotipo de la ES ya que, aunque sólo se han hallado en el 21% de los pacientes con ES (aparecen en mayor porcentaje en el síndrome de Sjögren o el LES), se asocian predominantemente con la EScl26.
VasculopatíaLas alteraciones vasculares son la base de las principales complicaciones sistémicas de la ES, incluyendo la HAP, la crisis renal de la esclerodermia (CRE) hipertensiva y la vasculopatía digital. El daño vascular se produce tempranamente en la ES, aunque su desencadenante es desconocido. Se desarrolla una alteración en la microcirculación (especialmente arteriolar) evidenciada por el daño estructural de los capilares del pliegue ungueal y por las respuestas vasoespásticas que se producen en el fenómeno de Raynaud27.
Se abren grandes espacios entre las células endoteliales, hay pérdida de la integridad de la capa endotelial y se produce vacuolización del citoplasma y apoptosis de las células endoteliales. Además de esto, aparece un infiltrado monocelular perivascular, se engruesa progresivamente la pared del vaso y aparecen lesiones obliterativas que llevan a la desaparición de los capilares. La escasez de pequeños vasos sanguíneos es característica de los estadios tardíos de la ES. A pesar de la pérdida progresiva de vasos sanguíneos y de los elevados niveles plasmáticos de factor de crecimiento del endotelio vascular, hay un defecto en la vasculogénesis, de mecanismo aún desconocido27. Existe también un déficit de los precursores de las células endoteliales circulantes, que además tienen dificultad para proliferar y madurar.
El 20–30% de los pacientes con ES tienen anticuerpos anticélulas endoteliales circulantes, que inducen la sobreexpresión de moléculas de adhesión en las células endoteliales y la apoptosis de éstas28,29. También se ha observado el depósito del complejo de ataque a la membrana (C5b-C9) en la microvasculatura de la piel dañada por ES, tanto en fases tempranas como tardías30.
Las endotelinas son potentes vasoconstrictores y tienen efecto fibrogénico, y desempeñan un papel importante en la ES. La secreción basal de endotelina-1 (ET-1) por parte de la célula endotelial supone un importante nexo precoz entre el daño de la célula endotelial y la activación de fibroblastos; esto será especialmente relevante en la afectación pulmonar y digital para el tratamiento31.
En respuesta a la inflamación y al daño vascular, habrá sobreexpresión de moléculas de adhesión, como ICAM-1, VCAM-1 y ELAM-1, tanto en circulación como en la superficie de las células endoteliales. Se unen a células de la serie blanca y a las plaquetas, con lo que se produce migración de estas células a través del endotelio hacia la matriz extracelular (MEC), lo que a su vez produce el citado infiltrado perivascular. La expresión de E-selectina en células endoteliales se correlaciona con el grado de infiltrado de células mononucleares en fases tempranas de la lesión de la ES, y tanto la E-selectina como el VCAM-1 podrían ser marcadores de progresión o regresión clínica de la ES32.
Las células mononucleadas que han migrado a la MEC expresan en su superficie marcadores de actividad, entre otros integrinas de clase β 1 y 2, que facilitan su unión a otras células como los fibroblastos y a componentes tisulares como el colágeno i y iv, la fibronectina y la laminina.
FibrosisEl depósito de MEC en exceso es causante en gran medida de la morbimortalidad de la ES. Los fibroblastos y los miofibroblastos activados depositan tejido conjuntivo fibroso, de modo que la fibrosis reemplaza gradualmente a la fase inflamatoria, modifica la arquitectura del tejido dañado y produce la mayoría de los síntomas27. En la piel, la fibrosis comienza en la dermis profunda y en la parte superficial del tejido celular subcutáneo (TCS), y aumenta a medida que desaparece la microvasculatura y se destruyen los anejos. Desde los estadios tempranos de la enfermedad parecen establecerse pequeñas subpoblaciones autónomas de fibroblastos que producen un exceso de MEC. Se ha observado que estas subpoblaciones se suelen hallar cerca de células inflamatorias mononucleares o adyacentes a vasos sanguíneos33. Son iniciadores de este proceso numerosas citoquinas y factores de crecimiento de múltiples vías de señalización34,35.
Los pericitos y las células musculares lisas son células presentes en los pequeños vasos. Los pericitos tienen la capacidad de transformarse en células musculares lisas de la pared vascular, fibroblastos y miofibroblastos, y participar en la proliferación de células endoteliales. El aumento del grosor de la pared vascular, producido por la proliferación de las células musculares lisas vasculares, indica que las células están respondiendo al daño inducido por la ES. Los pericitos de las lesiones sobreexpresan numerosos receptores de citoquinas, incluyendo el receptor del PDGF. Las células endoteliales son las únicas células mesenquimales que entran en apoptosis en la ES temprana, mientras que las células musculares lisas vasculares y los pericitos proliferan profusamente27.
Los fibroblastos son los encargados de la producción, el depósito y el remodelamiento de las fibras colágenas y de otros componentes de la MEC. Los fibroblastos en la ES se pueden convertir en miofibroblastos, y sobreexpresan diversas citoquinas (factor de crecimiento transformante β [TGF-β]) y receptores del TGF-β. Contienen también en la ES un exceso de especies reactivas de oxígeno (ROS). Estos fibroblastos tienen un defecto inherente en las microfibrillas que contienen fibrilina-1 y presentan sobreexpresión autónoma y mantenida del gen del colágeno tipo i36,37.
TGF-β. La isoforma 1 desempeña un importante papel en la fibrosis, ya que es un mitógeno indirecto de los fibroblastos, mientras que las isoformas 2 y 3 parecen tener efectos antifibróticos38. También es el más potente inductor de miofibroblastos y modula la expresión de varios receptores de citoquinas, incluyendo el propio TGF-β y el PDGF. Es causante de la elevada expresión del factor de crecimiento de tejido conectivo (CTGF), con actividad biológica similar al TGF-β. Una expresión aumentada de TGF-β39 y CTGF se ha detectado en lesiones iniciales de esclerodermia, pero después disminuyen los niveles del TGF-β y se mantienen elevados los del CTGF.
Inicialmente, el TGF-β se produce en células mononucleares del infiltrado inflamatorio, pero posteriormente lo producen los fibroblastos mediante regulación autocrina, en respuesta al TGF-β mediante la expresión de más receptores para el PDGF-α y el TGF-β1 en su superficie34. Un circuito autocrino del TGF-β podría explicar la activación persistente de la expresión de genes de colágeno en fibroblastos, y sería causante de la naturaleza progresiva de la enfermedad40.
Aunque la patogenia de la fibrosis sigue sin ser bien comprendida, hoy por hoy es evidente que la citoquina multifuncional TGF-β es esencial en el proceso. Existe una vía de señalización intracelular activada por el TGF-β; la transducción de señales comprende un sistema de receptores en la superficie celular y una familia de segundos mensajeros/factores de transcripción llamados Smads. Son proteínas evolutivamente conservadas. Los Smads son indispensables para la mayoría de las acciones profibróticas del TGF-β. Alteraciones en la actividad de los Smads conllevarían respuestas alteradas de la vía TGF-β con consecuencias catastróficas para el huésped41,42. Los niveles de Smad 3 (activadores) están elevados en fibroblastos de la ES, no así los de Smad 4 o 7 (inhibidores). Se desconoce el mecanismo por el que la vía del Smad de los fibroblastos está activada en la esclerodermia. La citada estimulación autocrina por parte del TGF-β endógeno podría ser una explicación43.
El PDGF está ligado a la cicatrización de las heridas y a la fibrosis. Hay presencia de anticuerpos estimuladores contra el receptor del PDGF en el suero de pacientes con ES. El PDGF se deposita en vasos sanguíneos de la dermis profunda en lesiones precoces de ES y estimula la transformación de pericitos en fibroblastos.
ET-1. Actúa conjuntamente con el TGF-β en la conversión de fibroblastos en miofibroblastos. Los efectos beneficiosos de los inhibidores de los receptores de la ET-1 sobre la HAP nos llevan a deducir su importancia en la patogenia de la ES. La interleucina-4 tiene efecto profibrótico en fibroblastos de la dermis de ES. Se han implicado numerosas citoquinas en la angiogénesis, la angiostasis, la fibrosis y la inflamación localizada de la esclerodermia, aunque aún sin resultados concluyentes sobre las vías en las que participan.
Las moléculas de colágeno de las lesiones de ES muestran enlaces cruzados habituales en las moléculas del hueso, pero no presentes normalmente en la piel. Las fibrilinas 1 son fibrillas producidas por fibroblastos de la ES, más inestables que las de personas sanas. La interacción entre TGF-β y fibrilina es necesaria para la activación de los fibroblastos en la esclerodermia. Las integrinas α y β subregulan la síntesis de colágeno por parte de los fibroblastos, mecanismo que falla en la ES.
ROS. Se han implicado niveles altos de ROS y de estrés oxidativo en la esclerodermia. En la mayoría de las enfermedades inflamatorias, los altos niveles de ROS son consecuencia directa de la activación de células sanguíneas mononucleares. En la ES parecen ser independientes del estado inflamatorio e inducen daño en el ADN. Además, los radicales libres tienen efectos profibrogénicos en los fibroblastos. El PDGF estimula la producción de ROS24.
Hipoxia. La hipoxia, frecuente en la piel de pacientes con esclerodermia, produce una activación de los fibroblastos, y una síntesis excesiva de ciertas citocinas estimuladoras, como el PDGF, el TGF-B1 y la endotelina. La transcripción de colágeno se ve aumentada cuando los fibroblastos se exponen a tensiones de oxígeno bajas, y esto puede deberse al TGF-β44.
Manifestaciones clínicasAunque el diagnóstico se establece sobre la base de hallazgos clínicos y serológicos, existen unos criterios diagnósticos propuestos en 1980 por la American College of Rheumatology que aún siguen vigentes45 (tabla 2). En estadios iniciales de la enfermedad, estos criterios no son útiles, ya que puede no haber afectación cutánea, por lo que se han postulado los siguientes hallazgos para facilitar el diagnóstico de una ES precoz o preesclerodermia: 1) fenómeno de Raynaud con amplias anomalías en la capilaroscopia del pliegue ungueal; 2) fenómeno de Raynaud con autoanticuerpos (ACA, antitopoisomerasa-i, anti-PM-Scl o anti-ARN-polimerasa i o iii)46 (tabla 4).
Criterios para la clasificación de la esclerosis sistémica (1980). American College of Rheumatology 2009
|
Las manifestaciones varían tanto en extensión como en gravedad, aunque hay ciertas características que se hallan de forma casi constante en estos enfermos, como el fenómeno de Raynaud y la esclerosis de la piel. Como hemos destacado en la introducción, la extensión de la afectación cutánea será la que marcará la diferencia entre un subtipo u otro (tabla 3). La EScd se caracteriza por la afectación cutánea del tronco y la parte proximal de las extremidades, con un fenómeno de Raynaud de pocos meses de duración, incluso posterior a la aparición de la clínica cutánea. En la EScl habrá afectación de la cara y de las zonas acras, con un fenómeno de Raynaud que precede en años a la aparición de la clínica cutánea. La EScl incluye el síndrome de CREST (figs. 1–4).
Clasificación de esclerosis sistémica
| Esclerosis sistémica cutánea limitada |
| Fenómeno de Raynaud de varios años de evolución |
| Afectación cutánea limitada a la cara, las manos, los pies, los antebrazos (distribución acral) |
| Capilares periungueales dilatados |
| Incidencia de hipertensión arterial pulmonar tardía (10–15%), con/sin calcificación cutánea, enfermedad gastrointestinal, telangiectasias (síndrome de CREST) o enfermedad intersticial pulmonar |
| Rara afectación renal |
| Anticuerpos anticentrómero |
| Esclerosis sistémica cutánea difusa |
| Fenómeno de Raynaud seguido de cambios cutáneos edematosos, de menos de un año de evolución |
| Afectación cutánea troncal y acral |
| Dilatación capilar periungueal, pérdida de capilares |
| Aparición temprana y significativa de afectación renal, pulmonar intersticial, gastrointestinal difusa y miocárdica |
| Anticuerpos anti-Scl-70 y anti-ARN-polimerasa-i, ii o iii |
CREST: calcinosis, Raynaud, esofagitis, esclerodactilia y telangiectasias.




La ES sin esclerodermia (tabla 4) es una rara variante de la enfermedad que se caracteriza por los típicos hallazgos vasculares y fibrosis visceral, pero sin esclerosis cutánea. El pronóstico se asemeja al de la EScl. Los anticuerpos más frecuentemente hallados han sido los ACA. Deberá cumplir los 4 siguientes criterios: 1) fenómeno de Raynaud o equivalente vascular periférico (úlceras digitales, capilares anormales en pliegue ungueal); 2) ANA positivos; 3) cualquiera de los siguientes: hipomotilidad esofágica distal, hipomotilidad de intestino delgado, fibrosis pulmonar intestinal, HTP (sin fibrosis), afectación cardíaca típica de esclerodermia, CRE, y 4) ninguna otra conectivopatía definida u otra enfermedad que explique los puntos 1, 2 y 347.
Otras formas de esclerosis sistémica
| Esclerodermia sin esclerodermia |
| Hipomotilidad esofágica distal, hipomotilidad del intestino delgado, fibrosis pulmonar intestinal, hipertensión arterial pulmonar (sin fibrosis), afectación cardíaca típica de esclerodermia, crisis renal de esclerodermia |
| Sin afectación cutánea |
| Fenómeno de Raynaud, úlceras digitales o capilaroscopia anormal |
| ANA positivos (antitopoisomerasa-1, ACA, o anti-ARN-polimerasa-i, ii o iii) |
| Sin otra enfermedad que explique los síntomas |
| Esclerodermia debida a agentes ambientales |
| Distribución generalizada de la esclerosis cutánea e historia de exposición a agente ambiental sospechoso |
| Síndrome de solapamiento (overlap) |
| Características de ES con otras de enfermedades reumáticas autoinmunes, como lupus eritematoso sistémico, artritis reumatoide, dermatomiositis, vasculitis o síndrome de Sjögren |
| Preesclerodermia |
| Fenómeno de Raynaud |
| Cambios en la capilaroscopia periungueal y evidencia de isquemia digital |
| Autoanticuerpos específicos circulantes –antitopoisomerasa-i (Scl-70), anticentrómero o anti-ARN-polimerasa-i, ii o iii |
ACA: anticuerpos anticentrómeros; ANA anticuerpos antinucleares; ES: esclerosis sistémica.
La esclerodermia inducida por agentes ambientales se caracteriza por la distribución difusa de la esclerosis cutánea combinada con la historia previa de exposición a un agente que haya podido precipitar la aparición de esclerodermia. Entre estos agentes se encuentran las resinas epoxi, tolueno, benceno, el cloruro de vinilo, los pesticidas y diferentes solventes orgánicos utilizados en las pinturas. También pueden producir este tipo de cuadros fármacos como la bleomicina, la pentazocina, el docetaxel o la metafenilendiamina. Del mismo modo, la ES se presenta en trabajadores de minas de oro y carbón. En los pacientes con silicosis, la probabilidad de desarrollar ES es casi 200 veces mayor que en una persona normal, es el llamado síndrome de Erasmus48. Parece que la exposición a la sílice produce la enfermedad en personas predispuestas y por mecanismos diferentes al desarrollo de silicosis49.
Los síndromes de solapamiento se tratan de afecciones en pacientes con datos de ES cutánea, que presentan también manifestaciones clínicas de otras enfermedades autoinmunitarias, como el lupus eritematoso, la dermatomiositis o la artritis reumatoide.
La ES puede derivar en la afección y el fallo grave de prácticamente cualquier órgano interno, y será el que marque el pronóstico de la enfermedad. Los riñones, el esófago, el corazón y los pulmones son los más frecuentemente implicados. La disminución de la motilidad esofágica es la afectación visceral más frecuente, y la pulmonar será la principal causa de muerte.
Síntomas generalesLa fatiga, el malestar, las artralgias y las mialgias son frecuentes en la ES, aunque con una prevalencia mal definida. El dolor que refieren los pacientes suele estar motivado por el engrosamiento de la piel, el dolor articular, el fenómeno de Raynaud y las úlceras digitales. También aparecen síntomas como pérdida de fuerza, depresión o dificultad para conciliar el sueño50.
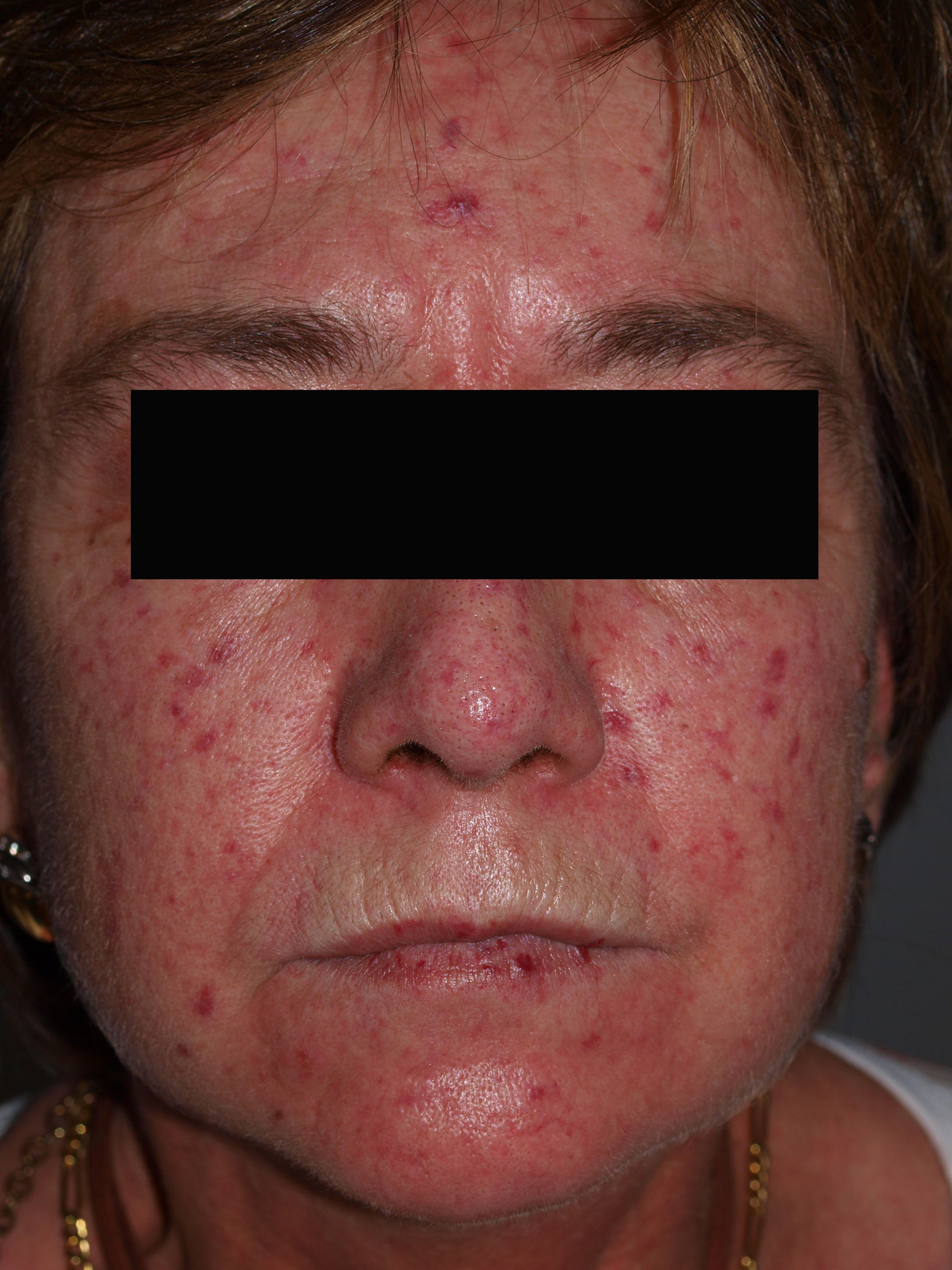
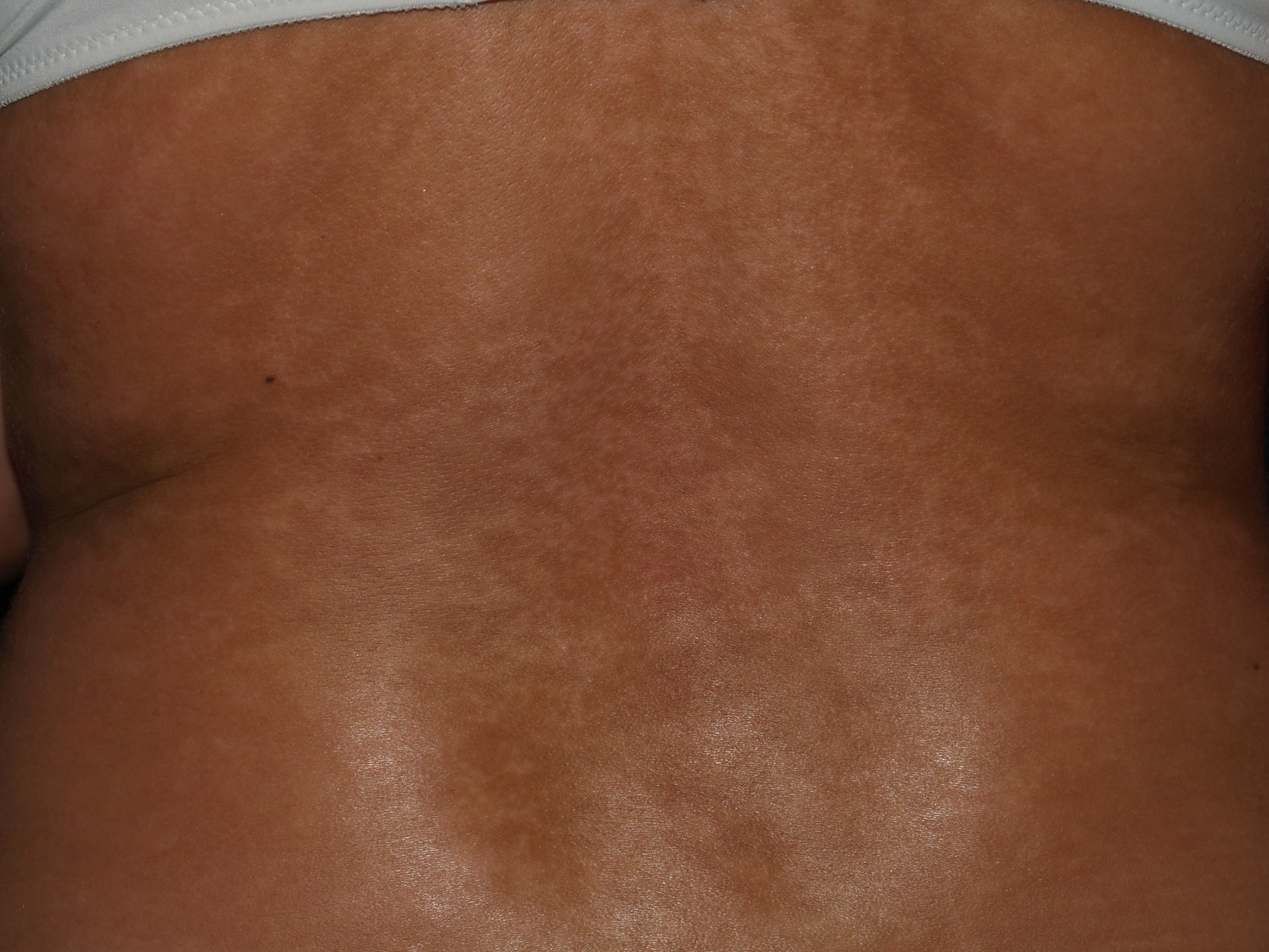
Manifestaciones cutáneasLa afectación cutánea es el hallazgo cardinal de la ES, y frecuentemente se inicia en los dedos y las manos. Se produce en 3 fases: edematosa, indurativa y atrófica. Comienza con hinchazón y tirantez en los dedos, más marcados por las mañanas, que normalmente no producen dolor. Tras un tiempo variable (más prolongado en el caso de la EScl), la piel poco a poco se engruesa y endurece. Se torna brillante, tirante y fuertemente adherida a los tejidos subcutáneos. La dermis se engruesa, pero la epidermis se adelgaza, con pérdida de pliegues y anejos y con la consiguiente pérdida de cabello y disminución de la sudación. Habrá cambios de coloración con hiperpigmentación/hipopigmentación (fig. 5), aparición de telangiectasias (fig. 4), presencia de una nariz afilada, microstomía y aspecto de máscara por pérdida de expresión de la cara. Finalmente, el engrosamiento de la dermis decrece y llega a quedar una piel atrófica. El sistema de graduación más utilizado es la escala de Rodnan modificada, que evalúa la esclerosis cutánea en 17 puntos diferentes, con baremaciones que van de 0 (normal), 1 (engrosamiento leve), 2 (engrosamiento moderado) a 3 (engrosamiento grave con incapacidad de pellizcar la piel de un pliegue); esta escala puede ser una herramienta útil y reproducible para el seguimiento de la historia natural de la enfermedad y la respuesta al tratamiento51–53.

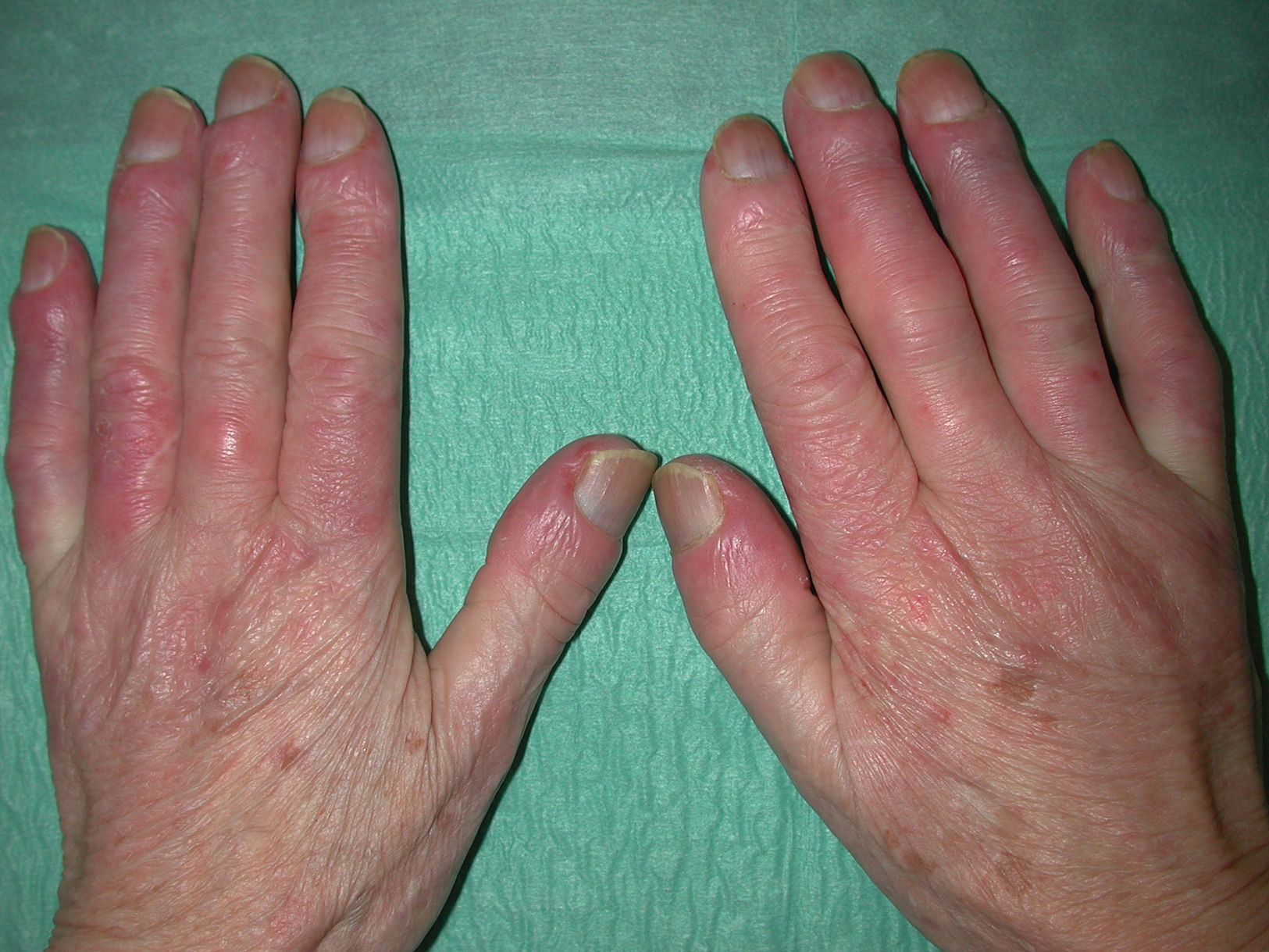
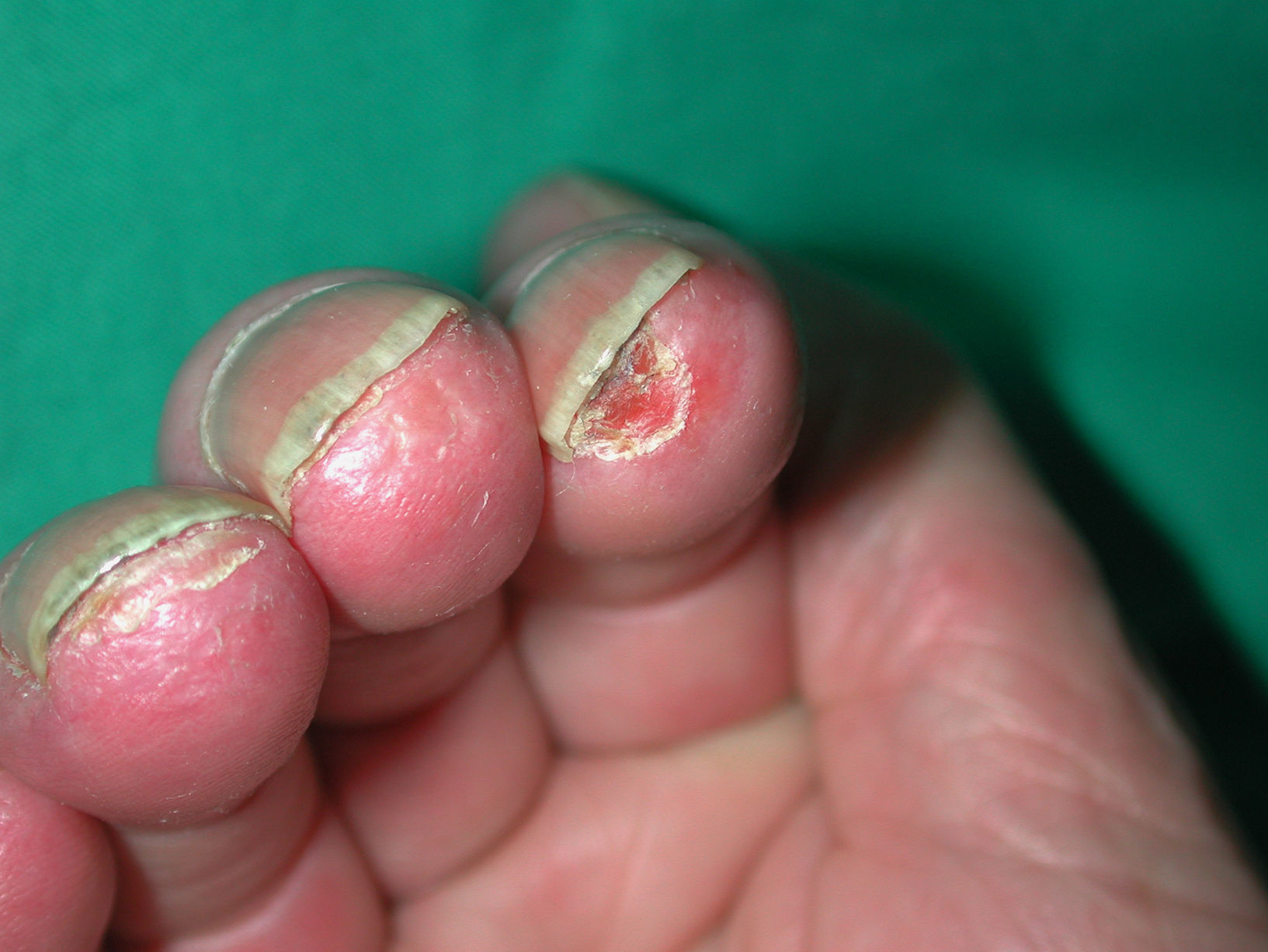
Úlceras digitales. Aparece en aproximadamente el 50% de los pacientes54. Su etiología es multifactorial, normalmente se producen por isquemia. Son más frecuentes en pacientes con ES y fenómeno de Raynaud que en aquéllos con fenómeno de Raynaud primario. Son más frecuentes en pacientes con EScd con afectación cutánea extensa, y producen dolor, sobreinfección, limitación funcional, resorción digital y osteomielitis (fig. 6). Existe más riesgo de que se desarrollen úlceras en pacientes con anticuerpo antifosfolípido positivo y presencia de áreas avasculares en la capilaroscopia55.

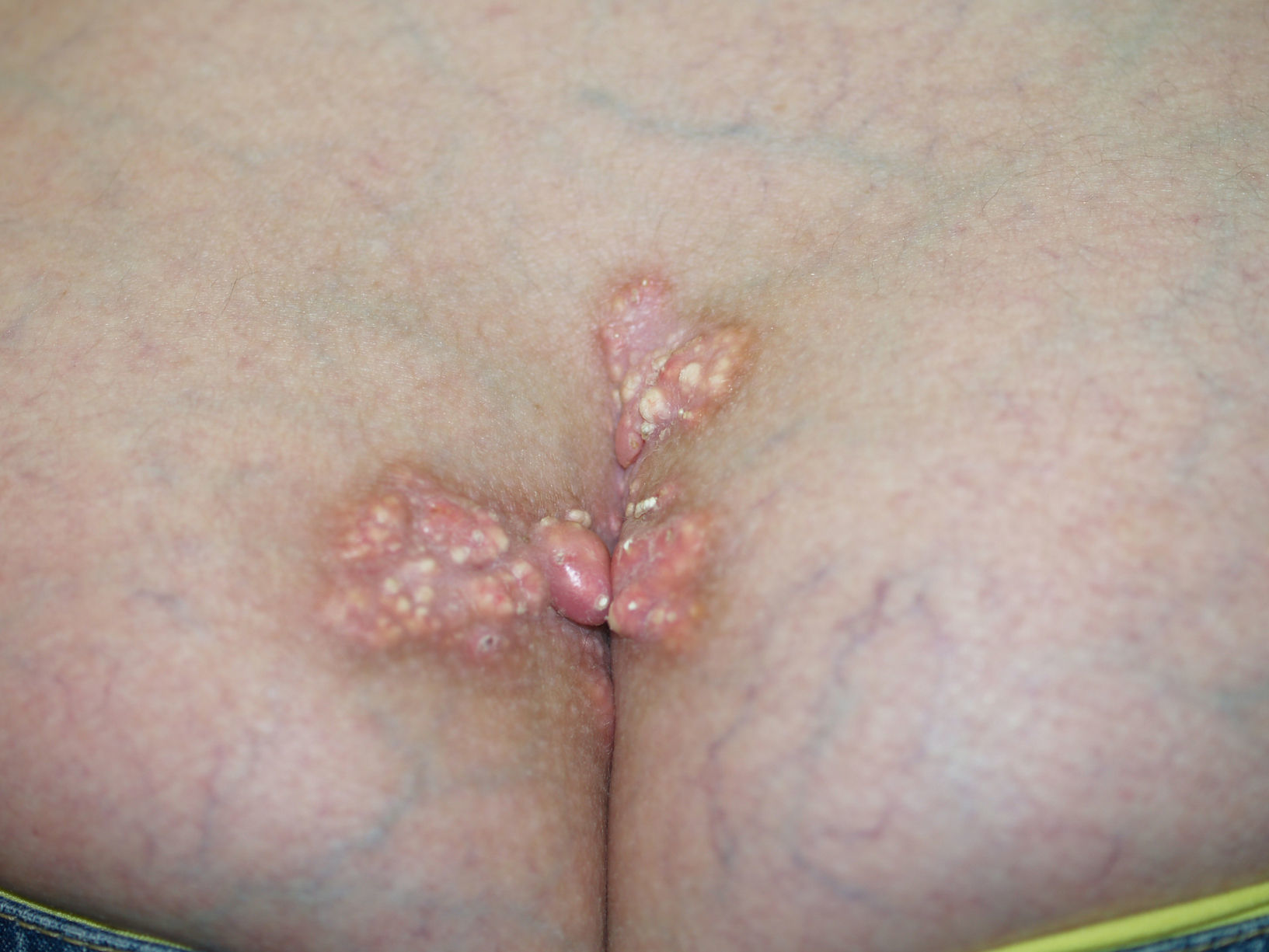
Calcinosis cutánea. Se producen depósitos en la piel o el tejido subcutáneo y varían de tamaño. El depósito se produce sobre todo en las almohadillas digitales y los tejidos periarticulares, al igual que en las zonas con traumatismos por presión (olecranon, rótula, etc.) (fig. 1). Pueden complicarse con ulceraciones.
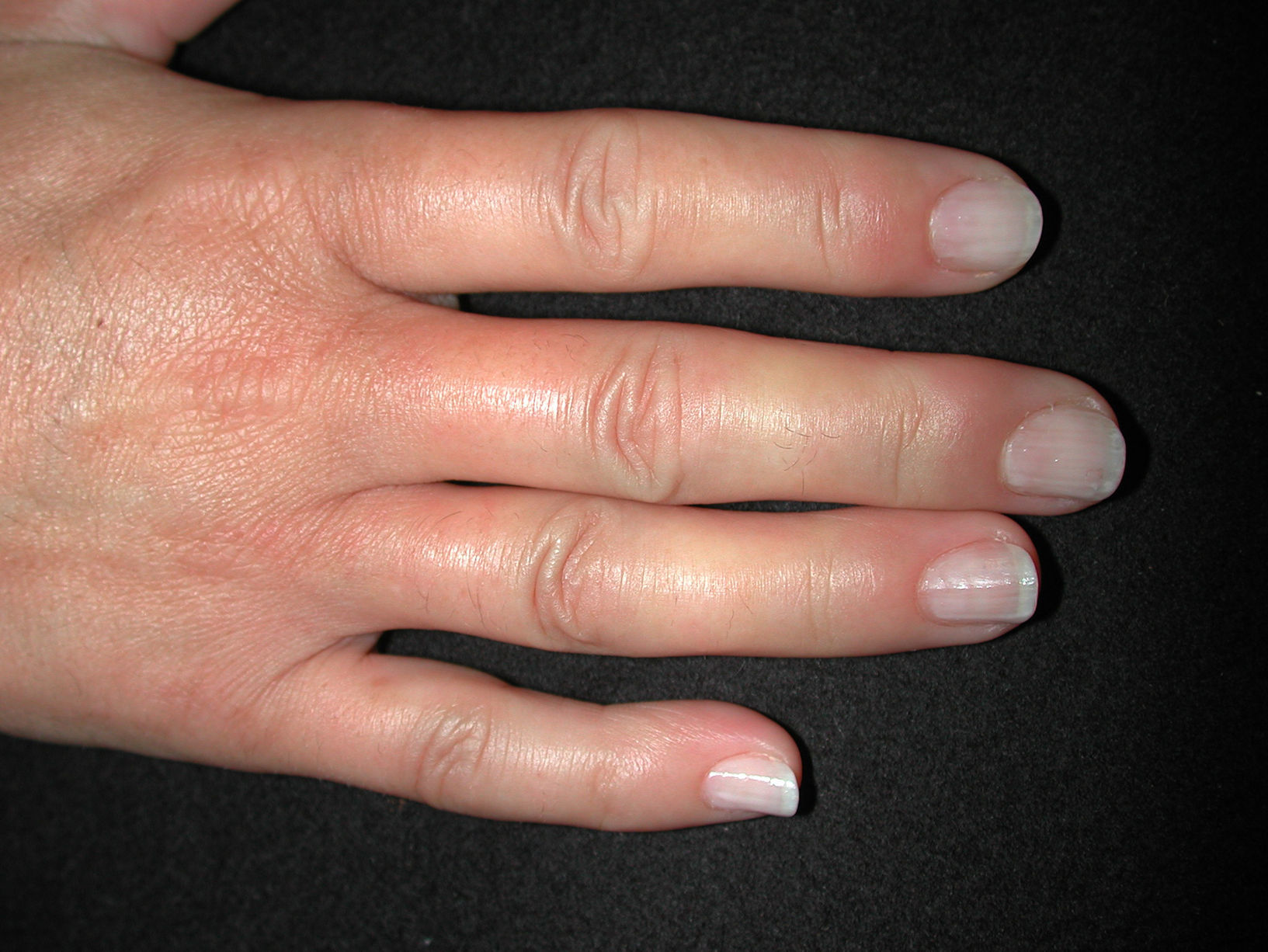
Fenómeno de Raynaud. Se define como el vasoespasmo paroxístico en respuesta a un estímulo emocional o por frío. Ocurre en el 95% de los pacientes con ES. Es un efecto trifásico que afecta a manos y pies, caracterizado por palidez brusca seguida de cianosis y, a veces, de eritema doloroso secundario a reperfusión vascular (fig. 2). Se debe a 3 mecanismos patogénicos: vasoconstricción, isquemia y reperfusión. Son frecuentes pequeñas áreas de necrosis isquémica en los pulpejos, que habitualmente conducen a ulceraciones. En la EScd sólo precede en meses al resto de síntomas; sin embargo, en la EScl lo hace durante años. Los pacientes con fenómeno de Raynaud aislado pero que también tengan autoanticuerpos de ES (ACA, antitopoisomerasa-i, o anti-ARN-polimerasa i o iii) deben considerarse como preesclerodermia56.
Manifestaciones musculoesqueléticasLos síntomas más frecuentes son debilidad muscular, artralgias y artritis. El dolor articular, la inmovilidad y las contracturas se deben a la fibrosis que está alrededor de los tendones y otras estructuras periarticulares. Hay autores que defienden que la palpación de una fricción en un tendón de un paciente asintomático puede ser predictiva de una afectación sistémica grave. Pueden aparecer miopatía (debilidad proximal leve, normalmente no progresiva) y miositis (solapamiento con polimiositis, pero es poco frecuente).
Manifestaciones gastrointestinalesEs la región más frecuentemente afectada tras la piel (entre el 75 y el 90% de los pacientes con ES). La alteración se centra en la fibrosis de la musculatura lisa intestinal. Se puede ver afectada cualquier parte del tracto gastrointestinal, desde la boca hasta el ano, aunque el esófago es el más implicado (el 80% de los enfermos57). Dada la atrofia de la musculatura lisa, habrá pérdida del peristaltismo esofágico, defecto del esfínter esofágico inferior e hipomotilidad gástrica, con los consiguientes problemas de reflujo gastroesofágico, esofagitis o esófago de Barret. También hay riesgo de aspiración pulmonar con la consiguiente producción/empeoramiento de la fibrosis pulmonar58. El colon y el intestino delgado se ven afectados con menos frecuencia, y esto ocasiona alteraciones de tipo malabsorción por sobrecrecimiento bacteriano, íleo intestinal con cuadros seudoobstructivos, estreñimiento, etc. Por último, también puede haber disfunción anorrectal con incontinencia fecal y prolapso rectal57.
Manifestaciones pulmonaresAfectación pulmonar. Aparece en más del 70% de los pacientes y en los últimos 15 años se ha convertido en la principal causa de muerte. La fibrosis pulmonar puede ser causa de una afección restrictiva progresiva grave, especialmente en aquellos enfermos con antitopoisomerasa-i. La HAP se observa en el 10–15% de los pacientes. Hay 3 patrones de afectación: personas con HAP aislada grave sin fibrosis pulmonar, que ocurre sobre todo en pacientes con EScl tras 10–30 años, en segundo lugar, HAP como complicación de fibrosis pulmonar intersticial, que se ve en enfermos con EScd, y en tercer lugar existe una afección mixta vascular y fibrótica, cuyo resultado es un proceso pulmonar indolente. Los síntomas observados son una disnea progresiva y, en estadios tardíos, síncopes. Puede haber derrame pleural o neumotórax espontáneo asociado a fibrosis grave o producirse una neumonía aspirativa como consecuencia del reflujo gastroesofágico59. Es importante señalar que hay un aumento de cáncer de pulmón en estos pacientes con ES, tanto fumadores como no fumadores. Por tanto, frente a una persona con fibrosis pulmonar grave de larga evolución que comienza con sintomatología nueva y grave, habrá que sospechar una neoplasia.
Manifestaciones renalesLa afectación renal puede manifestarse como una CRE hipertensiva. Se trata de la complicación más grave de la ES y la causa más frecuente de muerte hasta la introducción de los fármacos inhibidores de la enzima conversiva de la angiotensina (IECA). La crisis renal se produce en aproximadamente el 18% de los pacientes con EScd, y se presenta a modo de HTA acelerada y fracaso renal oligúrico. Pueden aparecer síntomas como cefalea, visión borrosa, disnea, microhematuria, aumento de creatinina, oliguria, anuria y anemia hemolítica microangiopática con trombocitopenia. Además de las crisis renales, aparecen alteraciones como HTA o elevación de creatinina hasta en el 50% de los enfermos. Estudios realizados en autopsias han demostrado que entre el 60 y el 80% de los pacientes con EScd tenía alteraciones renales. Aparecen durante los 4 primeros años de enfermedad, son casi exclusivos de pacientes con EScd y se han relacionado, entre otros, con el tratamiento corticoideo previo60,61.
Manifestaciones cardíacasLos pacientes con afectación cardíaca sintomática tienen peor pronóstico. Lo más frecuente es que se trate de afectación cardíaca secundaria a la hipertensión pulmonar, pero también se puede producir de forma primaria. Estas manifestaciones incluyen derrame pericárdico, fibrosis miocárdica y arritmias62.
Otras manifestacionesGenitourinarias. En el caso de los hombres, de un 12 a un 60% presentan disfunción eréctil. Las mujeres tienen disminución de la lubricación vaginal o estrechez del introito vaginal. Además, el 50% de las mujeres con ES tienen dispareunia.
Cabeza y cuello. Los síntomas más frecuentes son la xerostomía y la xeroftalmia. También hay dificultad para la abertura de la boca por la microstomía.
Neurológicas. Se cree que hay afectación neurológica hasta en el 40% de los pacientes. Puede haber anomalías en los pares craneales (más frecuentemente el trigémino y el facial) y en los nervios periféricos (neuropatía periférica, atrapamientos, etc.) o neuropatía autonómica (fenómeno de Raynaud, dismotilidad GI, arritmias cardíacas, impotencia). La mononeuropatía distal del nervio mediano es un hallazgo frecuente y precoz63.
Oculares. Es rara la participación ocular. Recientemente se han recopilado los casos publicados con afectación palpebral (tirantez, telangiectasias), conjuntival (congestión vascular, telangiectasias, queratoconjuntivitis seca) o retiniana (cambios epiteliales del pigmento retiniano, cambios hipertensivos). Son más raras las afecciones de la esclera o del tipo glaucoma64.
Neoplásicas. Existe un aumento de riesgo de tener cáncer, especialmente pulmonar, cutáneo y esofágico65 Aproximadamente un tercio de las neoplasias que se producen en la ES son pulmonares.
DiagnósticoSe establece principalmente con los hallazgos clínicos descritos. Es necesario realizar una detallada anamnesis y un cuidadoso examen físico.
El mayor grado de esclerosis cutánea se observa en las manos, sobre todo en estadios precoces de la enfermedad. La EScd puede ser difícil de diagnosticar, ya que en estadios iniciales predominan las artralgias y la hinchazón de tejidos blandos más que la esclerosis cutánea. La calcinosis o las telangiectasias pueden ser de gran ayuda en el diagnóstico, aunque pueden estar ausentes. La gravedad de la esclerosis de la piel puede evaluarse mediante la puntuación de Rodnan modificada51.
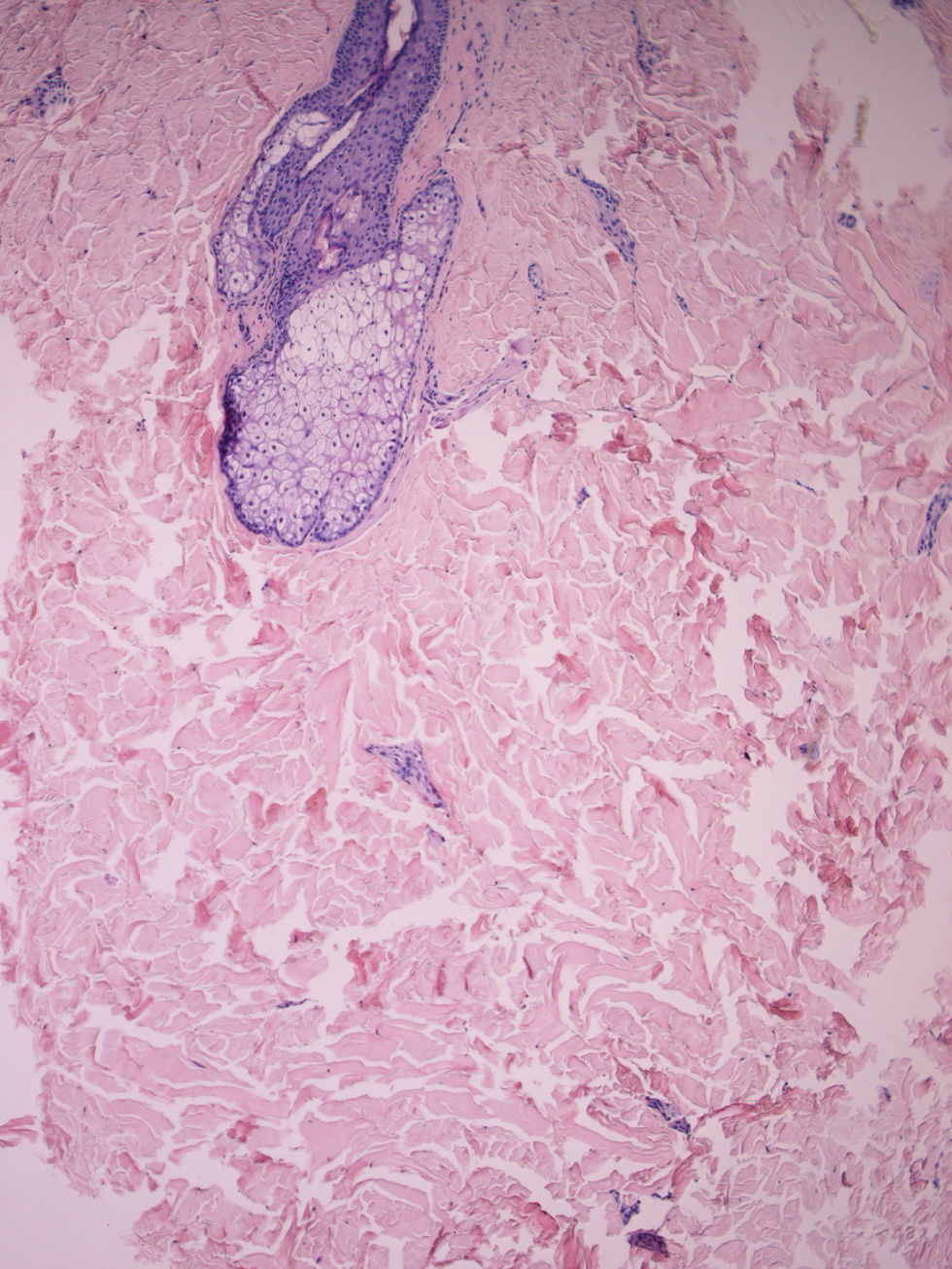
Biopsia cutánea. No suele ser necesaria, aunque puede ser útil a la hora de distinguir la ES de cuadros como la dermopatía fibrosante nefrogénica o el escleromixedema. En la anatomía patológica se observa un acúmulo excesivo de MEC, especialmente de colágeno tipo i y iii (fig. 7). En estadios precoces hay haces de colágeno engrosados en la dermis reticular, paralelos a la epidermis, e infiltrado inflamatorio entre los haces y alrededor de los vasos. El infiltrado puede extenderse al TCS y las glándulas sudoríparas. La epidermis suprayacente suele estar adelgazada y atrófica. Progresivamente va desapareciendo el infiltrado inflamatorio y se va volviendo avascular. En lesiones tardías se evidencia la esclerosis, las glándulas sudoríparas están atróficas o ausentes y el colágeno reemplaza a los adipocitos en el TCS53.

El estudio de los órganos internos y los análisis serológicos de los autoanticuerpos se realizan no sólo para diagnosticar, sino también para realizar una correcta clasificación. Se solicitarán radiografías de tórax, pruebas de función pulmonar, tomografía computarizada de alta resolución, lavado broncoalveolar o ecocardiograma para valorar la función respiratoria y cardíaca. Para descartar la afectación GI se realizará esofagograma, gastroscopia o manometría.
Capilaroscopia. Ha pasado a ser una herramienta útil tanto en el diagnóstico como en el seguimiento de la ES. Se han propuesto 3 patrones capilaroscópicos (precoz, activo y lento) que se correlacionan con la edad del paciente y la duración de la enfermedad y del fenómeno de Raynaud. En el patrón precoz hay presencia de pocos capilares dilatados o megacapilares (menos de 4/mm), pocas hemorragias capilares y distribución capilar bien conservada. En el patrón activo son frecuentes los capilares dilatados (más de 6/mm), las hemorragias capilares y una disminución moderada del número de capilares (20–30%). En el patrón lento se observan dilataciones irregulares de los capilares, ausencia de megacapilares y hemorragias, hay una disminución del 50–60% de los capilares, con grandes zonas avasculares y también hay numerosos capilares ramificados66,67.
Diagnóstico diferencialHay numerosos procesos que pueden tener una clínica similar a la de la ES, aunque la distribución y las características de la afectación cutánea suele ser «atípica». Normalmente carecen de afectación en los dedos y las manos, fenómeno de Raynaud, ANA específicos, alteraciones en la capilaroscopia o alteraciones organoespecíficas. A veces se encuentra el antecedente de exposición a agentes químicos, fármacos o coexistencia de algún otro proceso (neoplasia, infección, etc.). La biopsia cutánea puede ser de ayuda y deberá tener una profundidad suficiente. El diagnóstico diferencial se debe realizar con los siguientes cuadros cutáneos:
Esclerodermia localizada. Especialmente la morfea generalizada y la morfea en múltiples placas.
Escleromixedema. Caracterizada por pápulas cerosas con marcada esclerosis de la piel. Las manos, los brazos, la cara, el tronco y las piernas están afectadas. Es típica la paraproteinemia con predominio de IgG λ.
Síndrome POEMS. Acrónimo que recoge un complejo sintomático consistente en polineuropatía, organomegalia, endocrinopatía, gammapatía monoclonal, alteraciones cutáneas con hiperpigmentación local o generalizada; es raro fuera de Japon.
Escleredema. Enfermedad caracterizada por edema y rigidez cutánea rápidamente progresiva, que afecta especialmente a la parte alta de la espalda y el cuello, aunque pueden verse envueltos también los brazos, la cara y los hombros. Rara vez habrá afectación esofágica o lingual.
Dermopatía fibrosante nefrogénica. Afecta a personas de mediana edad con antecedentes de afectación renal. Se manifiesta por placas simétricas infiltrativas en el tronco, las piernas y los antebrazos. La cara normalmente está respetada.
Trastornos metabólicos. Como el mixedema, la porfiria cutánea tarda, las porfirias congénitas, la acromegalia y la fenilcetonuria.
Enfermedad injerto contra huésped crónica. Ocurre en el 30–50% de pacientes con transplante de médula ósea alogénico. La afectación cutánea puede ser localizada o generalizada. Normalmente no progresa de las manos, suelen aparecer pápulas liquenoides y placas poiquilodérmicas o lesiones tipo liquen escleroso y atrófico.
Otros procesos en los que aparece fenómeno de Raynaud como Raynaud primario, lupus eritematoso, artritis reumatoide, dermatomiositis y polimiositis.
TratamientoEl tratamiento de los pacientes con ES requiere no sólo una atención médica multidisciplinaria, sino también la colaboración del propio paciente, su familia, los fisioterapeutas, el personal de enfermería y terapia ocupacional, los trabajadores sociales y los servicios comunitarios53. Antes de iniciar el tratamiento, habrá que determinar qué afecciones orgánicas están relacionadas con inflamación o vasocontricción potencialmente reversibles (enfermedad activa) y cuáles presentan daño irreversible (como fibrosis o necrosis isquémica). Además hay que insistir en que el paciente abandone el hábito tabáquico si es fumador.
La European League Against Rheumatism Scleroderma Trials and Research group ha presentado una hoja de recomendaciones basadas en la evidencia para el tratamiento de la ES68. En este documento se incluyen 14 puntos (3 en relación con vasculopatía digital, 4 con HAP, 3 con afectación gastrointestinal, 2 con crisis renales, uno con enfermedad pulmonar intersticial y uno con afectación cutánea), todos ellos resumidos en la tabla 5.
Recomendaciones de la EULAR para el tratamiento de la esclerosis sistémica: documento del EULAR Scleroderma Trials and Research group (ESUTAR). 2009
Vasculopatía digital en relación a la esclerosis sistémica (fenómeno de Raynaud, úlceras digitales)
|
ES: esclerosis sistémica; EULAR: European League Against Rheumatism; HAP: hipertensión arterial pulmonar; PA: presión arterial.
Piel. Se han utilizado múltiples fármacos con diferentes resultados para disminuir/detener la esclerosis cutánea: esteroides tópicos, inhibidores de la calcineurina tópicos, corticoides sistémicos o inmunosupresores sistémicos (fármacos modificadores de la enfermedad) como el metotrexato (MTX), la ciclofosfamida, la ciclosporina A o la D-penicilamina.
Es bien conocido el efecto antiinflamatorio e inmunosupresor de la fototerapia con radiación ultravioleta A (UVA) o de la fotoquimioterapia con psoralenos (PUVA), por lo que suele ser uno de los tratamientos que se prueban y, ocasionalmente, da resultados satisfactorios. Parece que la radiación UVA inhibe la fibrosis e induce el ablandamiento de la piel esclerótica, especialmente en la EScl69.
Puede haber prurito intenso en las primeras fases de la enfermedad, donde el rascado y las excoriaciones pueden ser muy evidentes. No suele responder bien a los tratamientos disponibles, pero normalmente a medida que progresa la enfermedad el prurito cede. Pequeñas dosis de corticoides sistémicos junto con una correcta hidratación de la piel alivian parcialmente esta sintomatología.
Telangiectasias. Si suponen un problema cosmético, sobre todo las de la cara, se pueden camuflar con maquillaje o aplicar láser.
Ulceraciones cutáneas. Las ulceraciones, las grietas periungueales y la paroniquia son frecuentemente resultado de la isquemia y los traumatismos. En caso de infección, habrá que administrar antibióticos frente a Staphylococcus aureus durante al menos 2 semanas. Para su tratamiento se recomienda utilizar iloprost intravenoso, un potente vasodilatador que también inhibe la adhesión y la agregación plaquetaria, aumenta la deformabilidad de los hematíes y parece que favorece la reparación del endotelio dañado. También disminuye la producción de citoquinas profibróticas como el CTGF de los fibroblastos. Es eficaz para disminuir tanto la frecuencia como la gravedad de los brotes del fenómeno de Raynaud y parece que también puede tener efecto beneficioso sobre las úlceras digitales70,71. Se administra vía parenteral a dosis de 0,5 a 2ng/kg/min durante 6 horas al día, con una duración máxima de 4 semanas. Para prevenir la aparición de nuevas úlceras digitales está indicado el bosentan oral72,73, un antagonista no selectivo del receptor de la ET-1 (bloquea tanto el A como el B) que, sin embargo, no ha mostrado eficacia a la hora de curar las úlceras. Se administra vía oral, a dosis de 62,5 a 125mg al día.
Calcinosis. No hay ningún tratamiento médico que haya demostrado eficacia a la hora de erradicar o prevenir la calcinosis. Entre otros, se han utilizado el probenecid, la colquicina, los bifosfonatos o la warfarina, también bloqueantes de canales de calcio como el diltiazem74–76. En caso de lesiones grandes, la extirpación quirúrgica puede ser útil.
Tracto gastrointestinal. Los síntomas de reflujo gastroesofágico pueden tratarse con medidas básicas (elevación de la cabecera) y con inhibidores de la bomba de protones. La metoclopramida y la eritromicina son fármacos procinéticos también útiles. La estenosis esofágica puede requerir dilataciones endoscópicas periódicas y, en último lugar, intervención quirúrgica. El octeótrido es un análogo sintético de la somatostatina con el que se han obtenido buenos resultados para regular la motilidad intestinal y la incontinencia fecal77.
Pulmón. Habrá que realizar una correcta vacunación frente a neumococo y gripe. En el caso de la alveolitis o la fibrosis pulmonar, la ciclofosfamida puede ser efectiva. Para el tratamiento de la HAP se recomienda utilizar el bosentan, el sildenafilo (inhibidor de la fosfodiesterasa tipo 5) o el sitaxsentan (antagonista selectivo del receptor de la endotelina-A). En caso de afectación grave se administrará intermitentemente prostaciclina intravenosa (epoprostenol intravenoso). Con frecuencia se asocia anticoagulación y, para los estadios finales de la enfermedad, quedaría la opción de trasplante pulmonar.
Corazón. En el caso de la pericarditis se pueden administrar antiinflamatorios no esteroideos (AINE) o dosis bajas de corticoides. Para la miocarditis, sin embargo, se pueden administrar altas dosis de glucocorticoides, junto con digitálicos y diuréticos. En fases terminales se puede recibir trasplante cardíaco.
Riñón. El objetivo en estos pacientes es la detección precoz de la crisis renal. Así, los enfermos con EScd deben controlar su presión arterial diariamente y consultar ante aumentos de presiones sistólicas mayores de 30mmHg. Los IECA son los fármacos de elección; el captopril es el más utilizado, aunque un tratamientos intenso y precoz con otros antihipertensivos puede ser beneficioso (calcioantagonistas, alfabloqueantes o betabloqueantes). Si bien algunos pueden requerir diálisis, son raros los casos que precisan trasplante renal60,61.
Fenómeno de Raynaud. Se recomendarán como medidas básicas evitar la exposición al frío y utilizar guantes, además de abstenerse del tabaco y la cafeína. Existe una amplia variedad de tratamientos que sólo producen un alivio sintomático, no curan la enfermedad. Algunos fármacos utilizados para afecciones de otros órganos también tienen respuesta frente al fenómeno de Raynaud; se recomienda utilizar principalmente calcioantagonistas (nifedipino, felodipino, amlodipino) e iloprost intravenoso en los casos graves70,71. Los IECA, los bloqueantes de los receptores de la enzima conversiva de la angiotensina (losartan) o el sildenafilo también son útiles. La simpatectomía digital y la reconstrucción microvascular se han probado con éxito si existe afectación de grandes vasos.
Fármacos modificadores de la enfermedadTratamientos inmunomoduladoresCiclofosfamida: se trata de un agente alquilante citotóxico, indicado en caso de afectación pulmonar intersticial. Aunque los resultados de diferentes estudios son controvertidos, ha demostrado eficacia en la afectación cutánea en los casos de EScd. Además es beneficiosa en la función pulmonar administrada tanto por vía oral como intravenosa78,79.
Micofenolato de mofetilo: se han publicado casos en los que se ha obtenido beneficio tras su administración a modo de mantenimiento tras el tratamiento inductor con globulina antitimocítica80. Parece que es eficaz tanto en el tratamiento de la clínica cutánea como en la reducción de la progresión de la enfermedad pulmonar y tiene un buen perfil de seguridad81,82.
Metotrexato: además de mejorar la clínica cutánea, también estabiliza la progresión de la enfermedad pulmonar83. Actualmente es el tratamiento de elección en pacientes con síndromes de solapamiento tipo ES/miositis o ES/artritis inflamatoria84–86.
Azatioprina: la mayoría de los estudios publicados ha utilizado la azatioprina como taratamiento de mantenimiento tras la ciclofosfamida con buenos resultados79,87. Se trata de un fármaco en auge para el tratamiento de la ES, con un buen perfil de seguridad.
Tolerancia a colágeno humano tipo i: al utilizar colágeno bovino tipo i, se induce tolerancia al humano y se mejora de este modo la clínica cutánea. Hay resultados prometedores, aunque parece ser más útil en pacientes con EScd avanzada88.
Trasplante autógeno de células hematopoyéticas: parece que se obtienen buenas respuestas, aunque hay una importante mortalidad relacionada con el trasplante89. Son necesarios más estudios además de criterios de inclusión y protocolos de tratamiento.
Inmunoglobulina intravenosa: se ha utilizado a altas dosis y se han obtenido buenas respuestas90, aunque aún son necesarios ensayos controlados randomizados.
Plasmaféresis: en varios estudios se ha combinado la plasmaféresis91 con la inmunosupresión, por lo que es difícil determinar cuál de las 2 ha sido la causante principal de la mejoría de estos pacientes.
Clorambucilo, 5-fluoracilo: son agentes alquilantes con efecto inmunosupresor. No han demostrado eficacia.
Tratamientos biológicosAnti-TNF: con el etanercept se obtuvieron buenas respuestas en la clínica articular, pero no en la cutánea92. No se ha observado un claro beneficio al utilizar infliximab. Además, son frecuentes las reacciones infusionales por la posible aparición de anticuerpos antiinfliximab en muchos pacientes; es por esto que se ha propuesto administrarlo con un inmunosupresor como el MTX. De todas maneras, los anti-TNF no deben utilizarse de forma habitual, sólo en aquellos enfermos con marcada clínica inflamatoria o con síndromes de solapamiento84. El Rituximab asociado al MTX ha obtenido buenas respuestas93, no así utilizado solo.
Tratamientos antifibróticosD-penicilamina: agente quelante que bloquea los enlaces cruzados del colágeno. No se han observado diferencias al utilizar dosis altas o bajas94 y, aunque hay resultados controvertidos, los últimos estudios defienden su eficacia95.
Relaxina: es una hormona secretada durante el embarazo que produce relajación de los músculos pélvicos y remodelación del útero. También tiene efectos antifibróticos y antiinflamatorios. En los estudios de fase iii se ha demostrado que es ineficaz y que produce toxicidad renal con HTA grave, por lo que está contraindicada96.
Interferón γ: producido por las células T activadas. Activa los macrófagos e inhibe la síntesis de colágenos. Hay resultados prometedores97.
Interferón α: no aporta ningún beneficio en el tratamiento de la ES.
Futuro: factor de crecimiento transformante βA medida que se va comprendiendo el complejo entramado de la patogenia de la ES, van apareciendo nuevas dianas de posibles tratamientos. Los estudios más numerosos y prometedores se centran en el TGF-β y en el CTGF.
Imatinib mesilato: bloquea las tirosina cinasas, entre ellas el receptor del PDGF, y limita la proliferación de los fibroblastos dérmicos. Hay casos publicados donde ha sido beneficioso en la fibrosis nefrogénica sistémica por gadolinio y también en la ES98. Actualmente está en marcha un estudio prospectivo multicéntrico del imatinib en ES.
Anticuerpos anti-TGF-β (CAT-192): no parecen ser eficaces en la fase activa de la enfermedad, aún hay ensayos en fase i y ii99,100.
Anticuerpos anti-CTGF: secretados en respuesta a factores de crecimiento y otros estímulos como la hipoxia o el estrés, están sobreexpresados en enfermedades fibróticas. Estudios en modelos de fibrosis en ratones han demostrado que la fibrosis cutánea inducida por TGF-β puede inhibirse con un anticuerpo anti-CTGF101.
Agentes para prevenir el daño vascularEl epoprostenol y el treprostinil son análogos de prostaciclinas que han demostrado una mejoría de la supervivencia, pero con efectos adversos importantes102,103.
El bosentan es un antagonista mixto de la endotelina-A/B con la ventaja de administración oral. Corrige la HAP porque antagoniza la ET-1, un potente vasoconstrictor y mitógeno de músculo liso. Mejora la capacidad de ejercicio y la hemodinámica cardiopulmonar, disminuye las resistencias vasculares pulmonares y la presión arterial pulmonar. Es hepatotóxico, por lo que hay que ajustar las dosis.
El sildenafilo es un inhibidor de la fosfodiesterasa-5. Se administran 20mg vía oral 3 veces al día (Revatio®) produciendo preferentemente vasodilatación pulmonar. Disminuye el índice de resistencia vascular pulmonar y mejora el intercambio gaseoso en pacientes con fibrosis pulmonar grave e HAP secundaria. También, aumenta el flujo sanguíneo periférico en los enfermos con fenómeno de Raynaud. Los efectos secundarios son leves en las dosis utilizadas (cefalea, flush facial, congestión nasal y dispepsia). El tadalafilo es otro inhibidor de la fosfodiesterasa-5 específica del guanosín monofosfato cíclico (GPMc). Parece ser seguro y bien tolerado (20mg al día vía oral), y ha demostrado similar eficacia al sildenafilo para tratar el fenómeno de Raynaud104. Los IECA son eficaces en el tratamiento de la CRE, la principal causa de muerte en la ES antes de la utilización de estos fármacos.
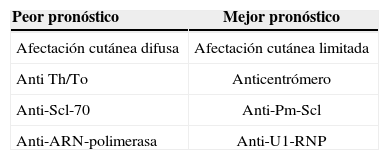
PronósticoLas tasas de mortalidad en los pacientes con EScd son 5 a 8 veces superiores que las de la población general, y es 2 veces superior en la EScl. En la EScl empeora el pronóstico la presencia de anticuerpos anti-Th/To. La supervivencia a los 15 años en el caso de la EScd será del 50%, mientras que en la EScl lo será del 70%. El 50% de las muertes se deben a HTP y el 25% a fibrosis pulmonar. De la totalidad de los pacientes con ES, el 50% que fallece por causas relacionadas con su esclerodermia lo hace por afectación pulmonar59. La afectación cardíaca confiere peor pronóstico54,105(tabla 6)
ConclusionesHasta este momento, la esclerodermia sigue siendo una enfermedad incurable, pero no por ello intratable. Asegurar el diagnóstico, establecer el subtipo, determinar el estadio evolutivo y, las posibles afecciones viscerales son los aspectos que se han de considerar antes de indicar el tratamiento más adecuado.