










Se describe el caso de una paciente de 25 años que presenta amenorrea primaria y esterilidad como consecuencia de la agenesia congénita de endometrio, sin otro tipo de alteraciones fenotípicas, cromosómicas o endocrinológicas asociadas. Se discuten aspectos de la fisiopatología, el diagnóstico y las posibilidades terapéuticas en la actualidad.
We report a case of primary amenorrhea and infertility due to congenital absence of the endometrium in a 25 year-old woman. There were no other malformations or endocrinological or chromosomal alterations. The physiopathological features, diagnosis and current therapeutic options in this malformation are discussed.
En ausencia de endometrio, la existencia de amenorrea es obvia. En los escasos casos publicados, la lesión es debida a una enfermedad adquirida, en general de origen traumático o inflamatorio producida, por ejemplo, tras un legrado vigoroso ya descrito por Asherman en 19481. El tejido endometrial también puede ser destruido como consecuencia de una endometritis tuberculosa o gangrenosa debida a una infección por gérmenes anaerobios2. Por último, se ha descrito la aplasia de endometrio secundaria a la administración de radio intrauterino sin que se altere la función ovárica.
Sin embargo, en la literatura no se encuentran con facilidad publicaciones acerca de casos de amenorrea primaria debida a ausencia congénita de endometrio sin que se asocie falta de desarrollo del resto de los tejidos del útero. Tampoco esta situación es específicamente descrita en los libros de texto de ginecología ni de endocrinología ginecológica. De hecho, en la revisión bibliográfica realizada, sólo se ha encontrado una publicación que hace referencia a esta extraña alteración, en la que se describen tres pacientes que fueron atendidas en Port Elizabeth (Sudáfrica) entre 1959 y 19603.
El presente trabajo pretende poner de manifiesto una etiología poco común de amenorrea primaria y discutir algunos aspectos de la fisiopatología, el diagnóstico y las posibilidades terapéuticas en la actualidad.
Caso clínicoMujer de 25 años de edad, de raza caucásica, remitida a la unidad de reproducción asistida de referencia por amenorrea primaria y deseo de gestación desde hacía dos años.
Los antecedentes familiares no mostraban ningún dato de interés, con ambos progenitores vivos y sanos. La madre había tenido cuatro gestaciones espontáneas que finalizaron en cuatro partos eutócicos de dos mujeres, y dos varones. La mayor de las mujeres era la paciente que ocupa este trabajo y la otra, de 21 años, había presentado una menarquia espontánea a los 13 años y tenía ciclos menstruales regulares.
La paciente nació a término tras gestación y parto normales. Su desarrollo psicomotor y pondoestatural en la infancia fue normal. No presentaba ningún antecedente médico de interés ni ninguna intervención quirúrgica. La telarquia y la pubarquia se produjeron normalmente a los 11 y 12 años, respectivamente. Inició relaciones sexuales a los 18 años, con su actual pareja, con la cual mantenía una media de dos relaciones coitales por semana sin dificultad. Refería que a los 23 años tuvo una deprivación escasa espontánea y desde los 18 años había sido tratada de forma ocasional con gestágenos y estroprogestativos orales para inducir deprivaciones sin ningún resultado.
Su pareja, de 26 años, no presentaba ningún antecedente familiar ni personal de interés. El espermiograma mostró parámetros seminales dentro de límites normales según criterios de la OMS.
La exploración física mostró una paciente de hábito normosómico y fenotipo femenino atlético. El peso era de 63,7 Kg, la talla de 1,75 m (IMC: 20,8). El perímetro de la cintura era de 75cm y el de la cadera de 89cm, con un índice cintura cadera de 0,8. La braza fue de 175cm. La tensión arterial fue de 100/60 mmHg y la frecuencia cardiaca de 88 lpm, rítmicos. La exploración mamaria mostró un desarrollo normal (Tanner 5), con mamas simétricas, sin evidenciarse galactorrea espontáneamente ni a la expresión. La pilificación era de distribución femenina y no presentaba hirsutismo. Los genitales externos y la vagina eran normales. El cérvix era ligeramente hipoplásico, de nulípara, bien epitelizado, sin eritroplasia periorificial. Se realizó una ecografía vaginal que mostró un útero hipoplásico de 40x35x21mm con una línea endometrial muy refingente de 2mm y con una longitud cervical mayor que la longitud del cuerpo uterino. Ovarios de 40x20mm, de aspecto poliquístico. El estudio de vascularización uterina realizada con Doppler pulsado mostró una vascularización miometrial escasa que disminuía en profundidad, hasta ser prácticamente inexistente en la proximidad de la cavidad uterina. El flujo arterial en arterias uterinas y a nivel miometrial presentó un índice de resistencia de 1 (figs. 1–4).




La analítica general fue normal y el cariotipo en sangre periférica fue 46,XX.
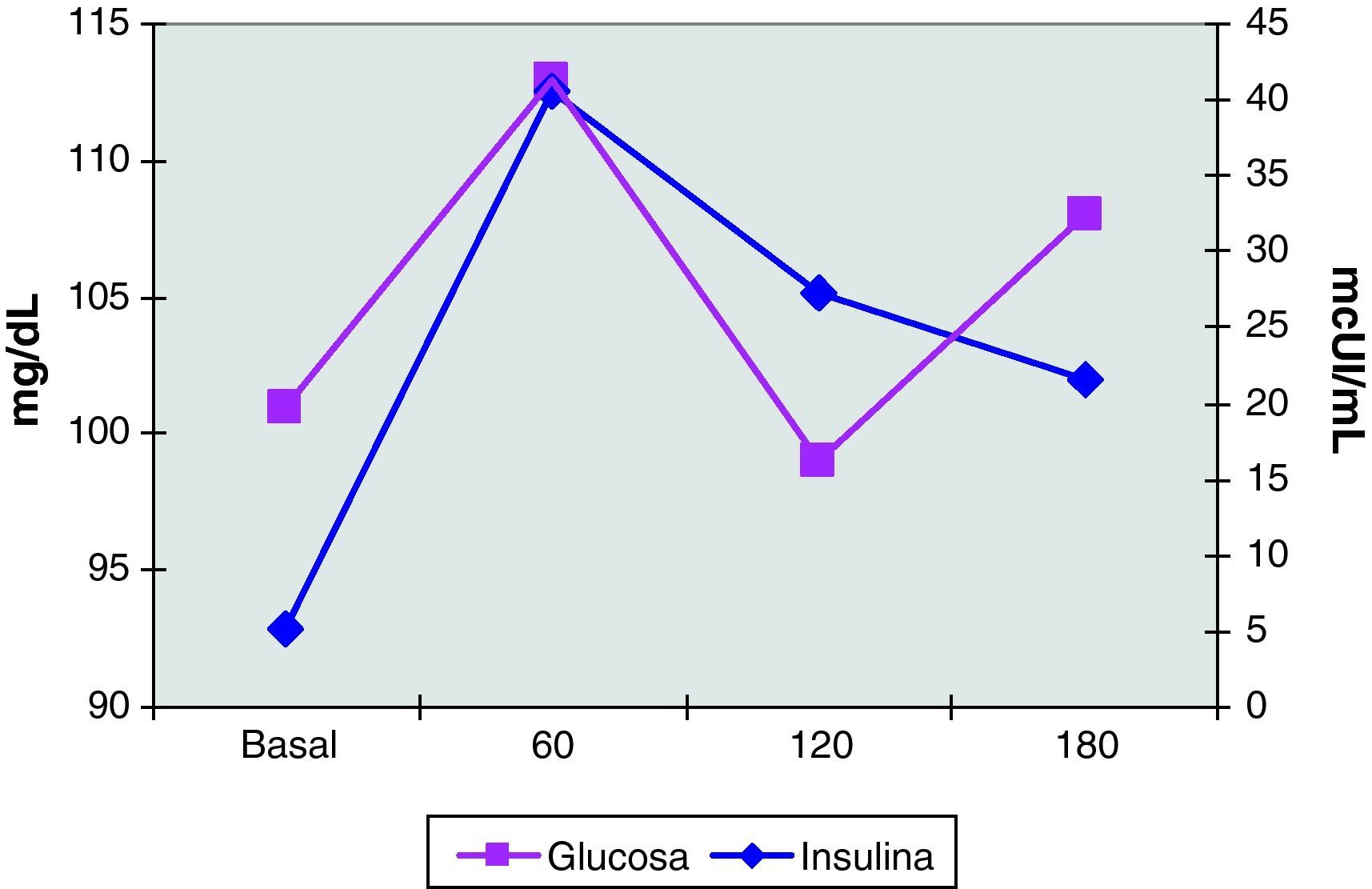
El estudio hormonal mostró niveles de FSH de 5 mUI/mL y de LH de 14,1 mUI/mL, con un cociente LH/FSH de 2,8. La prolactina fue de 19 ng/mL y la progesterona de 10,9 ng/mL. Las hormonas tiroideas eran normales. Se realizó un test de ACTH (determinaciones de cortisol, SDHEA y 17-hidroxiprogesterona basales y 6 horas después de la inyección IM de1mg de Nuvacthen depot) y una prueba de sobrecarga oral de glucosa con determinaciones de glucosa e insulina basales y a los 30, 60, 120 y 180 minutos de la ingesta de 75g de glucosa oral. Los resultados de dichas pruebas dinámicas fueron normales y se muestran en la tabla 1 y en la figura 5.
.")
Se realizó citología cérvico-vaginal con resultado normal. Se realizó un aspirado de cavidad uterina en el que sólo se obtuvo celularidad glandular endocervical sin evidenciar estroma ni glándulas endometriales, comprobándose la existencia de receptores de estradiol y progesterona en la muestra obtenida.
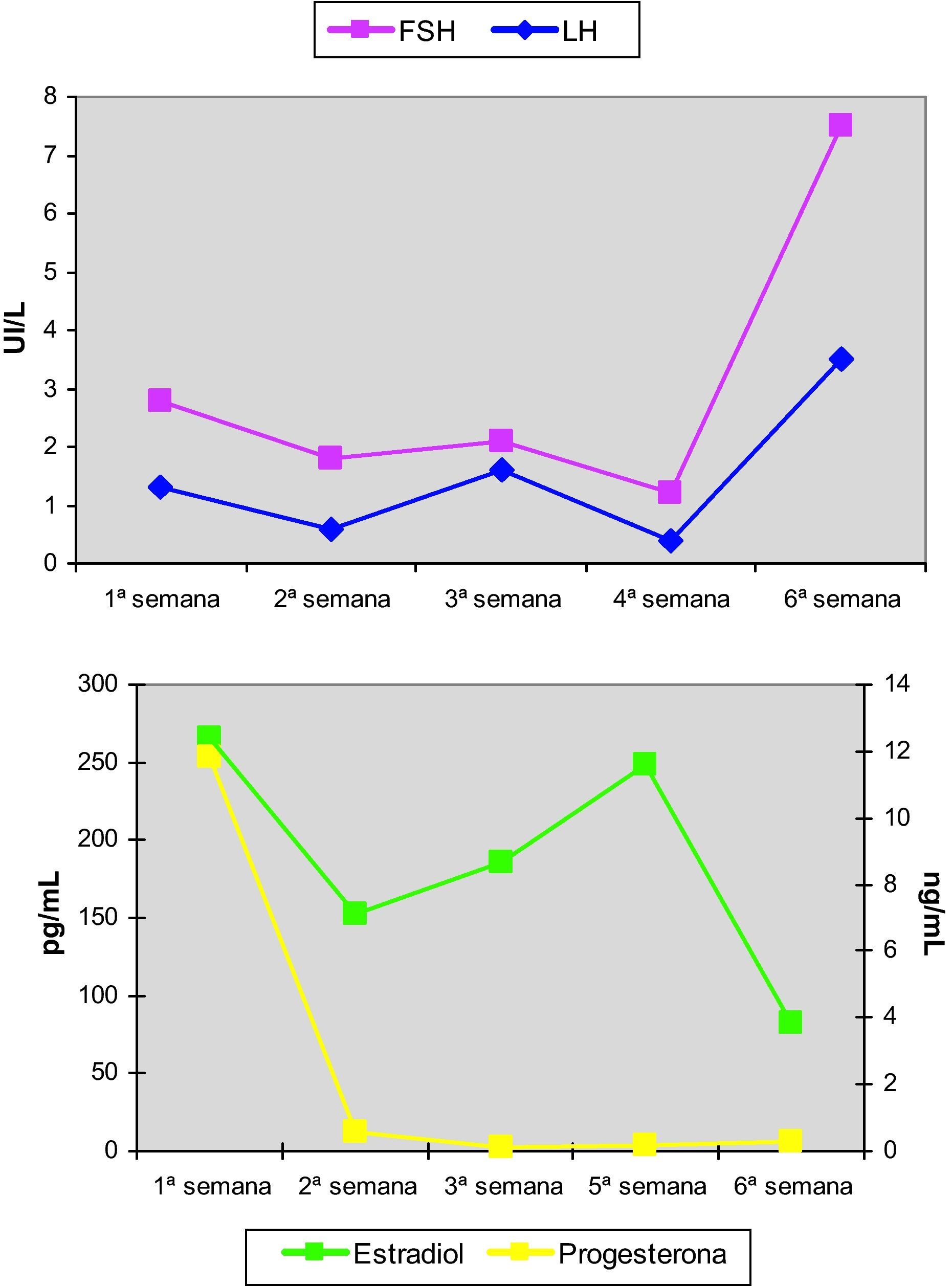
Se inició tratamiento con 17β-estradiol transdérmico a dosis progresivamente crecientes durante 5 semanas, practicando determinaciones hormonales y ecografías semanales. Los resultados de dichas determinaciones se muestran en la figura 6. El esquema terapéutico administrado fue:
- -
Primera semana: 1 parche transdérmico cada dos días de 6,4mg de 17β-estradiol (Evopad 100®, Lab. Janssen Cilag, Madrid). Cada parche libera 100μg/día de estradiol
- -
Segunda semana: dos parches transdérmicos cada dos días de 6,4mg de 17β-estradiol
- -
Tercera, cuarta y quinta semanas: tres parches transdérmicos cada dos días de 6,4mg de 17β-estradiol
- -
Sexta semana: dos parches transdérmicos cada dos días de 6,4mg de 17β-estradiol y 200mg/8h de progesterona micronizada vía oral (Progeffik 200, Lab. Effik, Madrid).

Tras la tercera semana de tratamiento, ecográficamente se observó una línea endometrial atrófica en tercio superior de cavidad uterina y de 6mm en istmo cervico-uterino. Se tomó una nueva muestra por aspirado de cavidad en la que se informó anatomopatológicamente de exclusiva representación de glándulas endocervicales con reactividad nuclear para receptores de estrógenos y progesterona, sin evidenciarse de nuevo representación glandular endometrial (fig. 7).

Al finalizar el tratamiento no se produjo ningún tipo de sangrado genital.
Con el diagnóstico de amenorrea primaria de origen uterino, se realizó una histeroscopia y laparoscopia diagnósticas con biopsia de cavidad uterina y ovario. En la laparoscopia se observó un útero pequeño, ambas trompas permeables normales y ovarios de aspecto normal (fig. 8). En histeroscopia se evidenció una cavidad tubular de 7cm de longitud con repliegue de aspecto polipoideo que se biopsió y resto de cavidad de aspecto atrófico. El estudio histológico del ovario mostró cortical ovárica de aspecto normal con folículos primarios. El estudio anatomopatológico de la muestra de cavidad uterina mostró pequeños grupos epiteliales glandulares de receptores estrogénicos y sin receptores de progesterona, sin identificarse glándulas endometriales (fig. 9).
Discusión
 y receptores de progresterona (RPg) (RE y RPg 250 x).")
Aunque el sexo gonadal viene determinado desde el momento de la fecundación, el desarrollo del tracto genital es indiferenciado hasta la semana octava de desarrollo. En este momento, los conductos mesonéfricos de Wolff y los paramesonéfricos de Müller están presentes como estructuras simétricas y pares. Estos, junto con el seno urogenital y los conductos metanéfricos, que formarán los uréteres en el individuo adulto, proporcionarán la fuente de tejidos que formará los genitales internos y el aparato urinario independientemente de las gónadas y de los riñones.
En la mujer, en ausencia de cromosoma Y, de testículos funcionantes y de hormona antimulleriana, los conductos de Müller no regresan y se desarrollan para dar lugar a las trompas de Falopio, al útero y la el tercio superior de la vagina, que se forman por la fusión de dichos conductos hacia la semana 10ª de gestación. La canalización posterior, que se completa en la semana 22, da lugar a la cavidad uterina, el canal cervical y la vagina. Bajo el epitelio se encuentra el tejido mesenquimatoso que dará origen al estroma uterino y a las células del músculo liso. En la semana 20 de gestación, la mucosa uterina está totalmente diferenciada en el endometrio4.
El endometrio deriva de la mucosa de los conductos de Müller fusionados y se considera uno de los tejidos más complejos del cuerpo humano, por su naturaleza cíclica y cambiante en respuesta a estímulos por parte de estrógenos y progesterona en el ciclo ovárico y a la interrelación compleja que existe entre sus propios factores autocrinos y paracrinos.
El inicio de la fusión de los conductos de Müller se produce en el centro del útero y posteriormente, la fusión progresa en sentido caudal y cefálico en la línea media. Las porciones no fusionadas en el extremo craneal se diferenciarán en las trompas de Falopio hacia la semana 10 de gestación. Después, se producirá la reabsorción del septo que une los dos conductos, que se inicia en el istmo y progresa en sentido craneal y caudal5. Se ha demostrado que la canalización del útero se produce después de la fusión6 y ha sido publicado un caso de amenorrea primaria por útero hipoplásico no cavitado7.
En resumen, la correcta diferenciación de los conductos de Müller en el tracto genital femenino depende de que se completen las tres fases de su desarrollo: organogénesis, fusión y reabsorción del septo.
La mayor parte de las malformaciones del tracto genital se explican por un fallo en la fusión de la porción inferior de los conductos de Müller, el desarrollo asimétrico de los dos, o la falta de desarrollo de uno o ambos ductos. Estas alteraciones anatómicas, sobre todo las debidas a la falta de desarrollo total o parcial de los conductos de Müller, pueden asociarse a otras malformaciones del tracto urinario, lo cual explica la íntima relación que existe entre los tres tipos de conductos durante los estadios iniciales del desarrollo.
Los genes HOX son una familia de moléculas reguladoras que controlan diversos aspectos de la diferenciación celular y la morfogénesis durante el desarrollo embrionario normal. Estos genes también se expresan en el útero adulto. Son esenciales para el desarrollo del tracto mülleriano durante el periodo embrionario y para la función del endometrio en la edad adulta y su expresión es regulada por esteroides sexuales, tanto en el periodo embrionario como en la edad adulta8.
Se ha demostrado en ratones que la exposición intraútero a dietilestilbestrol (DES) altera la expresión del gen HOX en el desarrollo del sistema mülleriano. En células humanas cervicales o endometriales, el DES induce la expresión de los genes HOXA9 y HOXA10 a niveles dos veces superiores a los inducidos por estradiol, y estas alteraciones explican el mecanismo molecular por el cual el DES afecta al desarrollo del tracto genital9.
Por otra parte, la exposición neonatal a DES afecta al desarrollo de las células del epitelio endometrial en ratones, de manera que, a los cinco días de vida, ratones expuestos a DES desde el nacimiento, mostraron alteraciones luminales y células epiteliales elongadas, que evolucionaron hasta presentar una total desorganización entre las capas estromal y miometrial a los 8 días y a una disminución en el número de glándulas endometriales con fibrosis estromal y cuernos uterinos más pequeños que los controles a los 22 días de edad10.
El caso que se presenta correspondería a una malformación mülleriana extraordinariamente rara y compleja en cuya etiopatogenia probablemente estaría implicado un defecto en la cavitación de los ductos tras una fusión completa y correcta. El diagnóstico diferencial habría que hacerlo con el útero unicorne o malformación clase II de la Sociedad Americana de Fertilidad, puesto que la cavidad era hipoplásica y de aspecto tubular. No obstante, esta alteración queda descartada, ya que en la laparoscopia eran perfectamente visibles y normales los dos cuernos uterinos y estaban presentes presentando una morfología normal las dos trompas de Falopio. Tampoco se evidenció ninguna imagen compatible con ningún esbozo de hemiútero atrófico o aberrante. Por otra parte, aunque el aspecto ecográfico de los ovarios y el la relación LH/FSH pudieran sugerir un síndrome de ovarios poliquísticos, la ausencia clínica y bioquímica de signos de hiperandrogenismo y la detección de niveles de progesterona típicos de ovulación, descartaron dicha alteración.
Aunque no sea posible de demostrar, al menos en este caso, el origen de la ausencia congénita de endometrio probablemente sea debida a la interposición de una noxa que afectaría a la normal expresión genética que daría lugar a la reabsorción normal del septo formado tras la fusión de los conductos de Müller y la cavitación posterior del útero, con el consiguiente recubrimiento de dicha cavidad por la mucosa endometrial, produciéndose todo ello alrededor de la semana 20 de gestación.
Los casos que se describen en la literatura corresponden a tres pacientes de 45, 24 y 22 años3. En las dos primeras, el diagnóstico de agenesia de endometrio se realizó a partir del estudio anatomopatológico del útero tras histerectomía abdominal. La tercera paciente, de 22 años, también fue diagnosticada tras la realización de una biopsia uterina de dos muestras que incluyeron la cavidad uterina en el curso de una laparotomía exploradora por amenorrea primaria. Debe matizarse que estos tres casos son publicados en 1961 y corresponden a pacientes que fueron diagnosticadas entre 1959 y 1960, con los métodos diagnósticos disponibles en aquel momento.
Aunque la prevalencia de este trastorno es muy baja, su diagnóstico debe sospecharse en cualquier protocolo de diagnóstico diferencial de amenorrea primaria en mujeres fenotípica y endocrinológicamente normales. En la actualidad, además de la ecografía, debe incluirse necesariamente la práctica de una histeroscopia y biopsia de cavidad y, siempre que sea posible, debería caracterizarse el aspecto morfológico del útero con una resonancia magnética o, mejor aún, con una laparoscopia diagnóstica, ya que resulta de gran utilidad para descartar otro tipo de defectos müllerianos.
Actualmente, el tratamiento resulta poco satisfactorio, fundamentalmente en lo que se refiere a sus posibilidades reproductivas. La única alternativa posible desde el punto de vista médico en el momento sería la subrogación, no permitida legalmente en nuestro país. Hipotéticamente, o al menos en teoría, podría ensayarse el desarrollo de líneas celulares que fueran capaces de diferenciarse en células endometriales para realizar un trasplante. No obstante, hasta el momento, los intentos de transplante de células endometriales en úteros desprovistos de este tejido no han producido el resultado esperado.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.