


La agenesia lumbosacra es una malformación congénita muy infrecuente que forma parte del síndrome de regresión caudal.
Presentamos un caso diagnosticado como agenesia lumbosacra en nuestra consulta de diagnóstico prenatal durante el segundo trimestre de gestación. No se presentaron malformaciones asociadas, el cariotipo fetal fue normal y la gestante carecía de antecedentes de interés.
La pareja se acogió al derecho de interrupción legal del embarazo.
Lumbosacral agenesis is an uncommon condition which is part of the caudal regression syndrome.
We report a case de lumbosacral agenesis detected by ultrasound in the second trimester of gestation. The fetus was not presenting other malformations. The fetal karyotype was normal and the pregnant woman did not have precedents of interest. The parents decide to abort.
La secuencia de regresión caudal comprende un conjunto de malformaciones lumbosacras que afectan a las extremidades inferiores y los esfínteres, generalmente asociadas a otras anomalías. No existen datos consistentes sobre la incidencia global de este síndrome, aunque la incidencia de la sirenomielia se estima entre 1:60000-1:100000 nacidos vivos1.
Los defectos estructurales observados en este patrón de malformaciones son variados e incluyen desde la simple ausencia de cóccix hasta la ausencia de toda la columna a partir de la 12 vértebra torácica.
Presentamos un caso clínico de agenesia lumbosacra dentro del espectro del síndrome de regresión caudal (SRC).
Caso clínicoPaciente secundigesta, de 21 años de edad, sin antecedentes médicos personales ni familiares de interés.
Como antecedente quirúrgico presentaba una laparotomía con apendicectomía y la extirpación de un quiste paramesonéfrico realizada en 2004.
No presentaba antecedentes familiares de interés. Tuvo un parto eutócico a término en 2005.
Se realizó la primera ecografía a las 13+1 semanas; se dató la gestación que correspondía a 11+1 semanas por CRL. Se repitió a las 2 semanas con una medida de translucencia nucal de 1,5mm.
No se realizó el cribado combinado por determinación de PAPP-A y fracción libre de gonadotropina coriónica humana beta (β-HCG) anterior a las 8 semanas de gestación.
Los resultados del triple cribado fueron normales, tanto para el cálculo del riesgo de síndrome de Down, como para los valores de alfafetoproteína en suero materno.
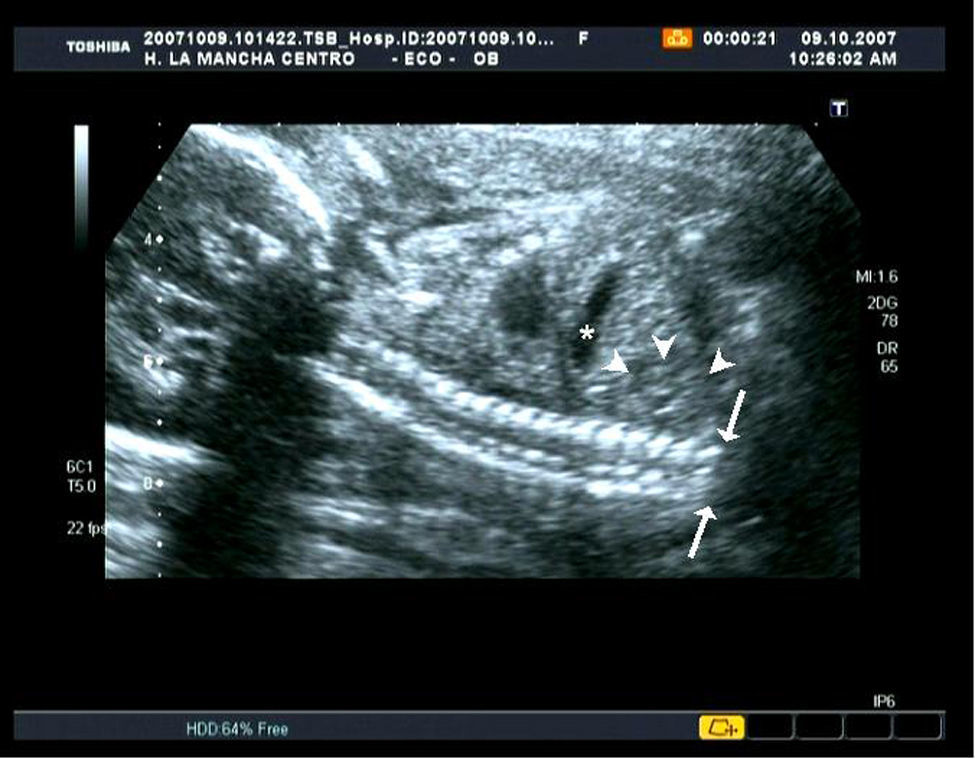
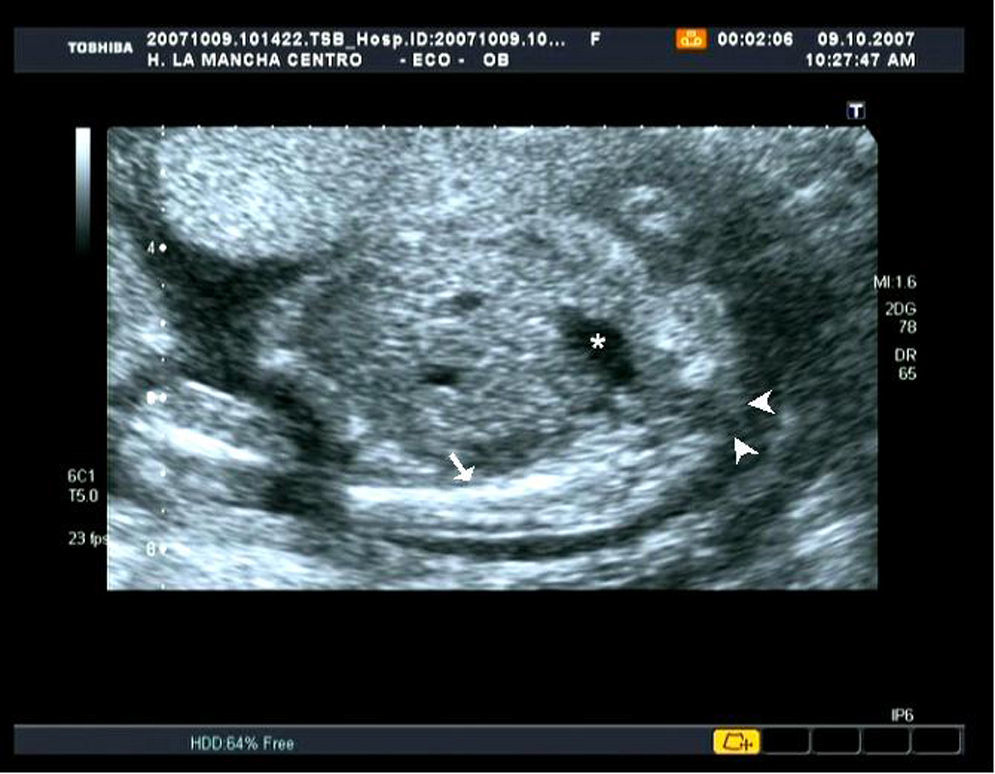
En la ecografía morfológica se realizó una biometría que resultó acorde con datación inicial de la gestación. Como hallazgos patológicos se encontró una ausencia parcial de vértebras lumbares y total del sacro (figs. 1 y 2). Se visualizaron ambos riñones y la vejiga de características normales. Los miembros inferiores se presentaron individualizados y con un tamaño acorde con edad gestacional, aunque su disposición era anómala y la movilidad estaba muy disminuida.


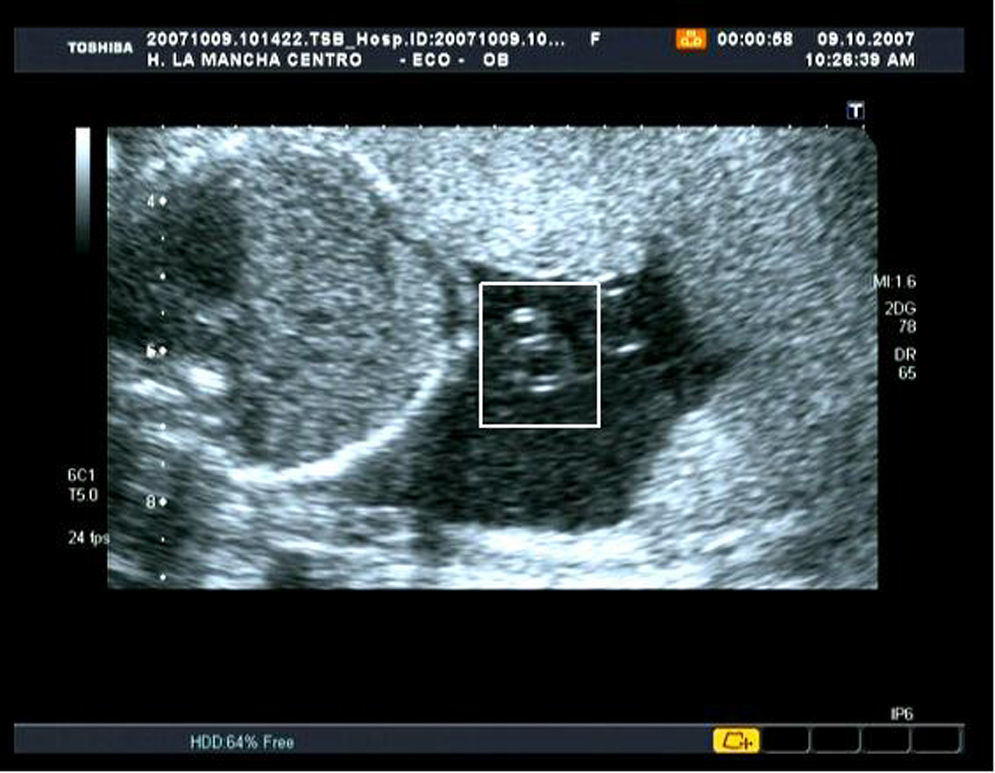
Se evidenció ausencia de defectos de tubo neural, cardíacos y gastrointestinales evidentes por ultrasonografía. Se apreció líquido amniótico en cantidad normal y destacó la presencia de un cordón con arteria única (fig. 3).
.")
Se realizó una amniocentesis genética, con el resultado de alfafetoproteína normal en el líquido amniótico, acetilcolinesterasa negativa, y cariotipo 46XX sin anomalías estructurales.
Con el diagnóstico definitivo de agenesia lumbosacra, la gestante solicita una interrupción legal del embarazo.
DiscusiónEl término de SRC fue propuesto por primera vez por Duhamel2, en 1961, para describir un espectro de malformaciones que puede variar desde la existencia de defectos aislados en el desarrollo del sacro, con ausencia parcial o total de este y defectos de su segmentación, hasta el grado más severo, constituido por la fusión de los miembros inferiores, conocido como sirenomielia.
Este autor postula que el origen de este síndrome es un defecto primario del mesodermo que determina la falta de inducción de un número determinado de somitas caudales del embrión, que permite la fusión de los primordios de los miembros, con ausencia o desarrollo incompleto de las estructuras caudales intercurrentes extendiéndose a varios niveles craneocaudales3. El resultado es un espectro de malformaciones congénitas donde se presentan alteraciones esqueléticas (agenesia lumbosacra) combinada con deformidades variables de los miembros inferiores y malformaciones del tracto digestivo, como la agenesia anal o ano imperforado, genitourinario, cardiopatías congénitas, fisura palatina y defectos del tubo neural. La alteración neurológica derivada de la afectación de la médula espinal en su porción distal ocasiona incontinencia y déficits motores en miembros inferiores4,5.
Las diferencias fundamentales que se han descrito entre la sirenomielia (a la que durante mucho tiempo se consideró una entidad independiente del SRC) y el resto de anomalías incluidas en el espectro de este síndrome, es que en estas últimas se visualizan dos extremidades inferiores hipoplásicas, las anomalías renales no suelen ser letales, el ano puede estar imperforado o no, el líquido amniótico aumentado o normal y el cordón presenta dos arterias umbilicales6. Nuestro caso cumple los criterios comunes al resto de SRC, ya que no presenta agenesia renal, fusión de las dos extremidades o reducción del líquido amniótico, a excepción de la normalidad del cordón umbilical, en el que encontramos una sola arteria (fig. 3).
No se conoce con certeza el factor desencadenante de los defectos en el blastocisto que dan origen a este síndrome. Las teorías que plantean los diferentes autores son la existencia de una diabetes materna, la predisposición genética y la hipoperfusión vascular7,8. Aunque el SRC es de presentación esporádica, en el 16-22% ocurre en hijos de madres diabéticas9. Presenta un bajo riesgo de recurrencia a pesar de que se han descrito patrones de herencia autosómicos recesivos y dominantes10 con una mayor incidencia de espina bífida oculta en los progenitores4. En el caso descrito no se encontraron factores relacionados, ya que la gestante presentaba cifras de glucemia dentro de normalidad, y no existían antecedentes familiares ni personales de interés.
La técnica de elección para el diagnóstico prenatal de este conjunto de malformaciones es la ultrasonografía. Aunque existen casos descritos de diagnóstico en el primer trimestre, son aquellos que presentan el cuadro más grave y letal del espectro, como es la sirenomielia y que son fácilmente identificables en la ecografía de las 11-14 semanas, tanto por la existencia de una única extremidad inferior como la alteración de líquido amniótico, aunque en este período gestacional no se presenta un oligoamnios grave11,12. Lo habitual, fuera de los casos comentados con anterioridad, es que el diagnóstico se realice durante la ecografía morfológica del segundo trimestre, que permite además una valoración del resto de malformaciones asociadas. En algunos casos descritos en la literatura científica, el diagnóstico se confirmó mediante ecografía 3D y resonancia magnética11.
En nuestro caso, realizamos el diagnóstico en la ecografía del segundo trimestre donde se visualizó una agenesia lumbosacra (figs. 1 y 2), con extremidades anómalas en su posición y movimiento pero no en su tamaño. No se apreciaron malformaciones renales, gastrointestinales, cardíacas, faciales ni del sistema nervioso central. No se pudo descartar ano imperforado ni fisura palatina, ante las dificultades de diagnóstico por imagen que presentan esas patologías.
Aunque el síndrome no está asociado a aneuploidías, es aconsejable la realización del cariotipo como en cualquier otra malformación fetal12. El cariotipo fetal informó de que se trataba de una niña cromosómicamente normal. También fue normal la determinación de alfafetoproteína en líquido amniótico, que nos confirmó la ausencia de un defecto de tubo neural abierto asociado.
Existen diversas clasificaciones de las agenesias sacras según las características radiológicas posnatales de las malformaciones y su intensidad; la frecuencia de las anomalías viscerales asociadas es muy similar, con independencia de la gravedad de la alteración ósea. El caso descrito pertenece al grupo I de la clasificación de Bollini13, que engloba los casos de agenesia sacra total con agenesia lumbar o toracolumbar de altura variable. Los problemas ortopédicos normalmente asociados son luxación de cadera, rodillas en flexo y pies zambos irreductibles que imposibilitan la deambulación.
Otras alteraciones previsibles en el caso descrito son las secundarias a la disrupción de la médula espinal. Incluirían defectos sensoriales y motores de los miembros inferiores, así como incontinencia de esfínteres4.
Una vez informada, la pareja se negó a proseguir estudio y solicitó la interrupción legal del embarazo.
Se debe destacar, por tanto, la importancia de un diagnóstico temprano de este tipo de malformaciones, a pesar de su baja frecuencia, ya que nos permite, por un lado, dar la opción a la pareja de que se acoja legalmente a una interrupción voluntaria del embarazo y, por otro, en el caso que desee proseguir con la gestación, poder planificar el nacimiento en las condiciones más adecuadas y con la participación de un equipo multidisciplinar que dependerá, a su vez, del tipo y alcance de los defectos.