



Se presenta el caso de una persona adulta de 24 años con genitales externos ambiguos, nunca estudiada ni tratada y que se había etiquetado de hermafroditismo en su país de origen. Estos casos son evaluados en su mayoría al nacimiento, siendo el diagnóstico más frecuente el de hiperplasia suprarrenal congénita y uno de los más infrecuentes el de la disgenesia gonadal parcial 46XY que presentamos.
We report the case of a 24-year-old adult with ambiguous external genitalia, who had never been studied or treated, but who was labelled as a hermaphrodite in the country of origin. These cases are usually evaluated at birth, the most frequent diagnosis being congenital adrenal hyperplasia. We present a case of one of the least common of these disorders, 46XY partial gonadal dysgenesis.
El término disgenesia gonadal se utiliza para designar el desarrollo anormal de la gónada fetal. La alteración gonadal puede asociar alteraciones en el desarrollo de los genitales internos y externos1.
Incluye diversas patologías, entre las que se encuentran el síndrome de Turner y sus variantes, la disgenesia gonadal completa 46XX o 46XY, la disgenesia gonadal parcial (DGP) 46XY o 45XO/46XY y el hermafroditismo verdadero.
Caso clínicoPaciente de 24 años, de nacionalidad rumana, etiquetada en la infancia de hermafroditismo pero nunca estudiada ni tratada con terapia hormonal.
AnamnesisAntecedentes familiares no valorables: padres fallecidos en accidente de tráfico Antecedentes personales: sin interés, salvo amenorrea primaria.
ExploraciónPeso: 65 kg; talla: 180cm, e índice de masa corporal: 20,1.
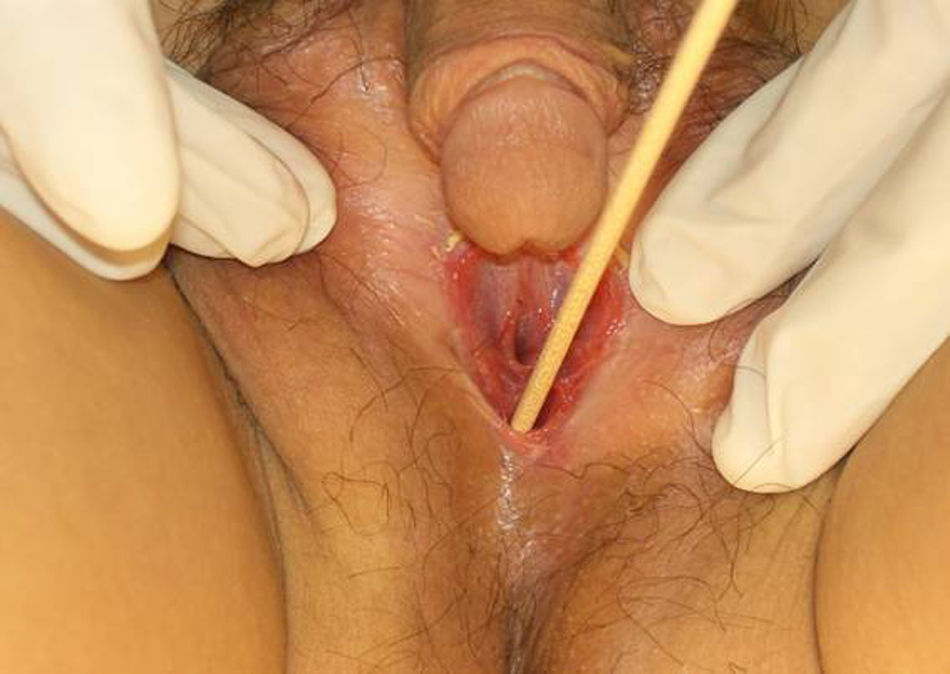
Presenta genitales externos ambiguos, con clítoris peniforme de 3 x 4cm, labios mayores atróficos fusionados con hendidura vaginal visible de 5mm y una longitud de 6-7cm explorada con torunda (fig. 1).

No existe desarrollo mamario (fig. 2) ni vello axilar, torácico o facial y el vello púbico presente es ralo (fig. 3).
Exploraciones complementarias

- -
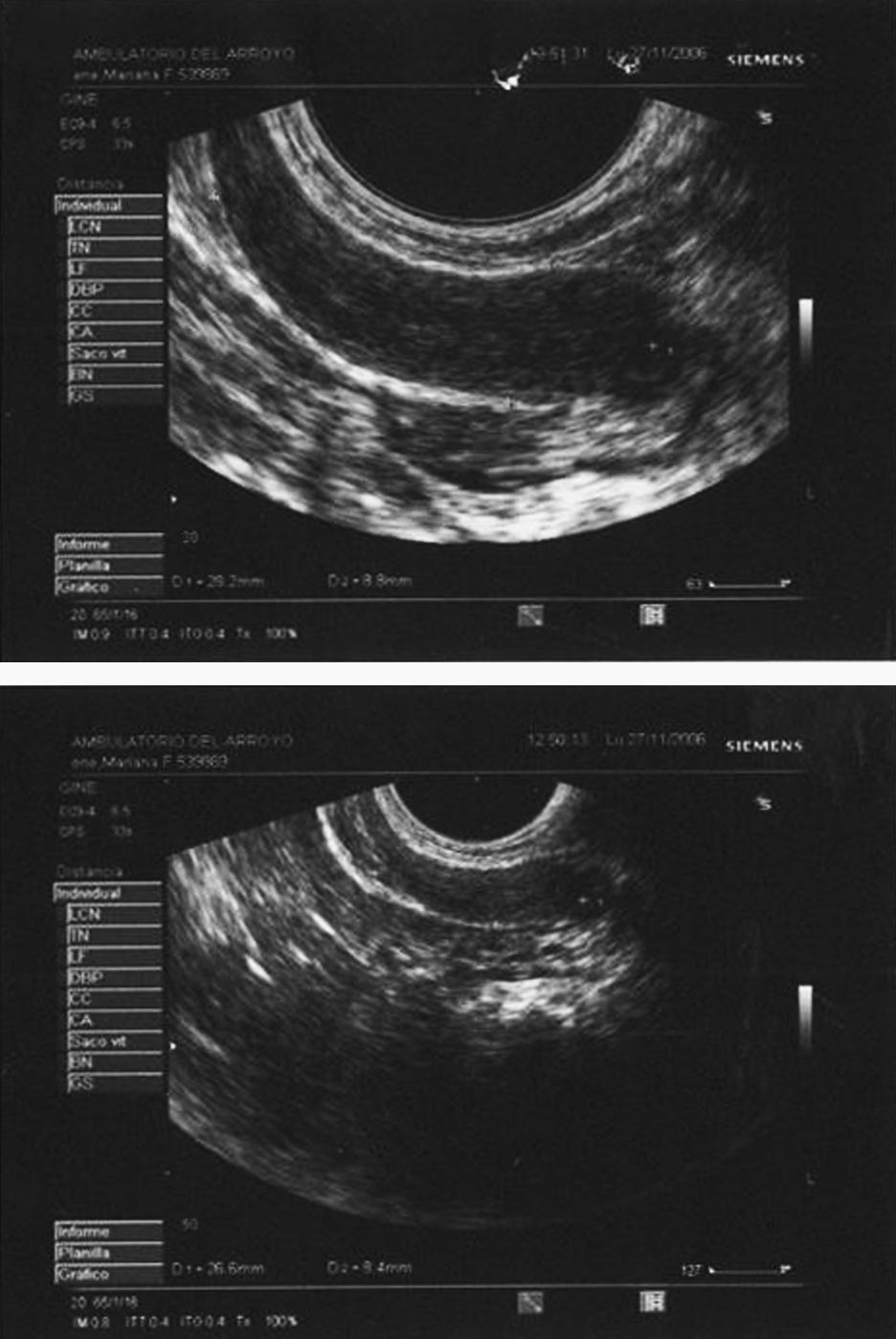
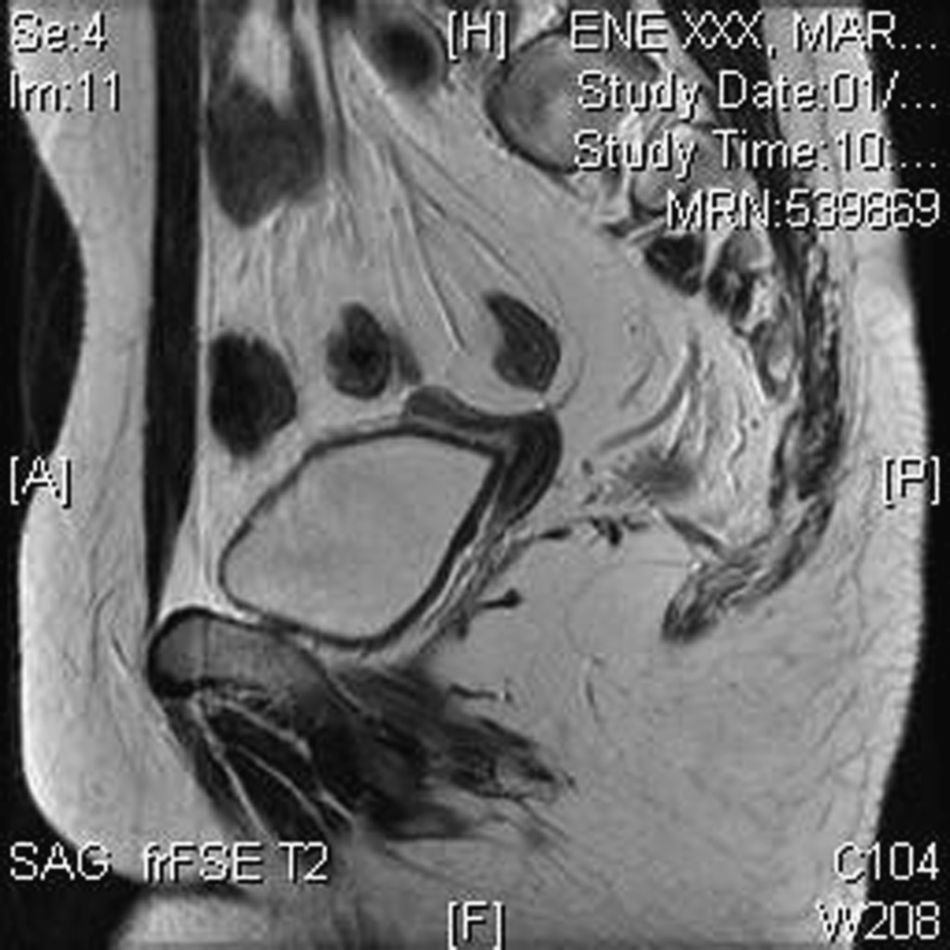
Ecografía transrectal: sospecha de útero rudimentario. Histerometría 28 x 10 x 8mm. No se visualizan gónadas (fig. 4).
- -
Analítica: compatible con insuficiencia gonadal (hipogonadismo hipergonadotropo), descartándose alteración suprarrenal2.
- -
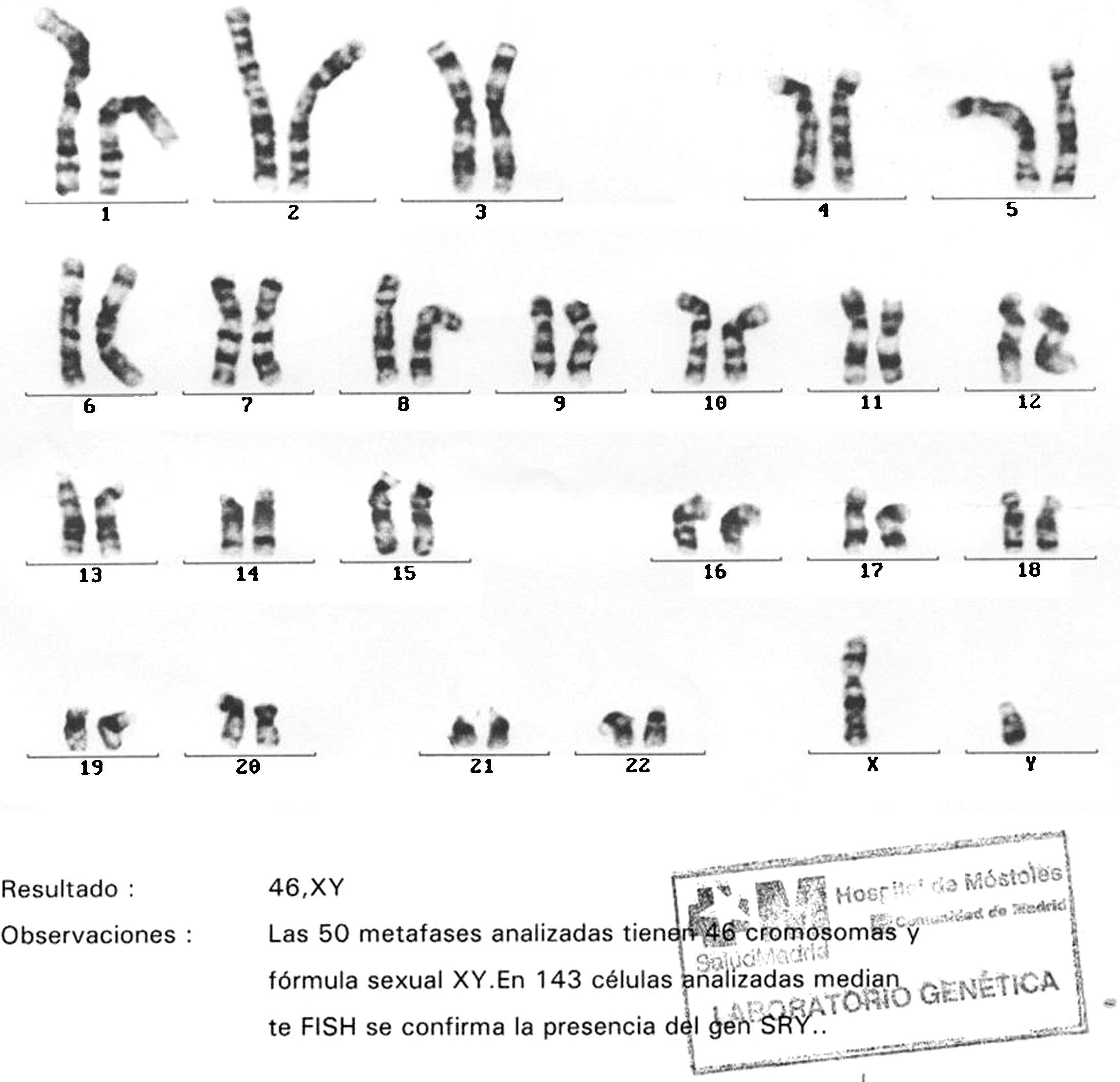
Cariotipo: 46XY. (Las 50 metafases analizadas tienen 46 cromosomas y forma sexual XY. En 143 células analizadas mediante FISH se confirma la presencia del gen SRY; fig. 5).
- -
Aporta informe de salud mental remitido desde el servicio de genética de referencia para valoración de identidad sexual no concluyente por falta de experiencia en ese terreno; la paciente se identifica como varón y vive como tal desde su permanencia en España.
- -
Resonancia magnética de pelvis con hallazgos superponibles a los ecográficos: se visualiza posible rudimento uterino de 2cm y la revaluación tras la cirugía permite identificar los rudimentos gonadales (fig. 6).
- -
Ecografía abdómino-pélvica y de partes blandas: sin alteraciones significativas, no se visualizan útero ni anejos en la pelvis. Se exploran ambos trayectos inguinales, sin imágenes nodulares que sugieran presencia gonadal, apreciándose en ambos lados tejido hipoecogénico alargado en relación con grasa en el trayecto del cordón.



Con el diagnóstico de sospecha preoperatorio de disgenesia gonadal XY, se realiza una laparoscopia diagnóstica con los siguientes hallazgos.
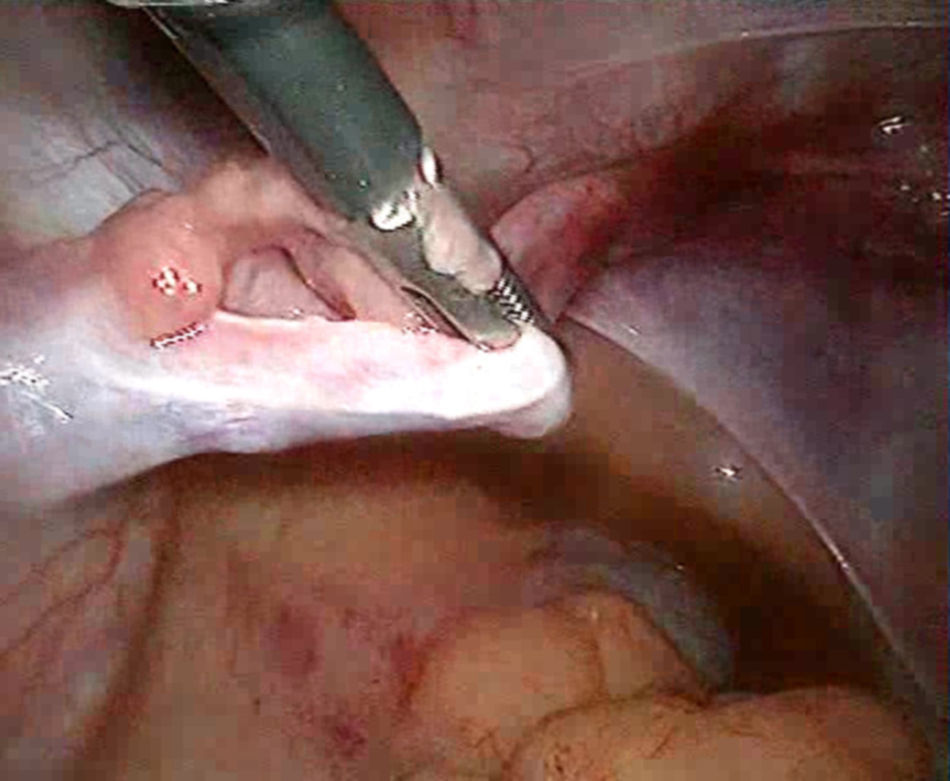
Se visualiza un repliegue vesical y un fondo de saco de Douglas sin evidencia de útero; se comprueba, con torunda y tacto rectal combinado, un fondo de saco ciego en el abdomen.
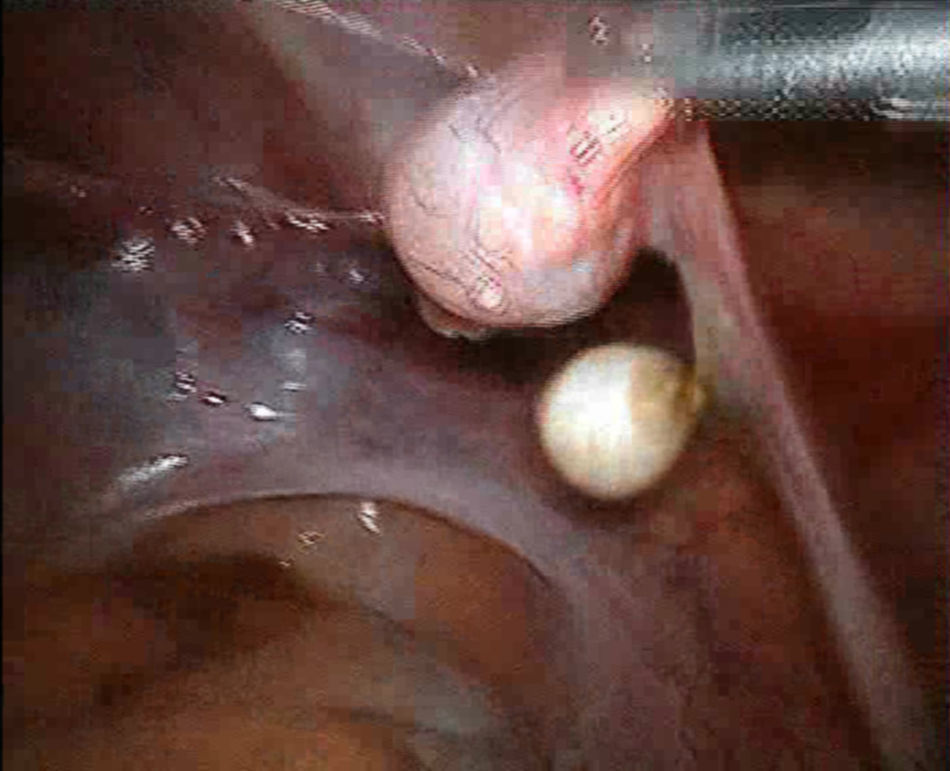
A nivel de la pared lateral se visualizan restos anexiales; el derecho con gónada redondeada de 1cm, de superficie nacarada y trompa atrófica, con porción ampular en maza, y el izquierdo con trompa atrófica y cintilla (figs. 7 y 8).


Se realiza una anexectomía bilateral por el riesgo de malignización de las gónadas disgenéticas, con presencia del cromosoma Y con el resultado anatomopatológico de:
- -
Anejo derecho: trompa uterina sin lesiones relevantes. Quiste paratubárico derecho. Testículo con fibrosis y atrofia tubular con calcificaciones múltiples. Túbulos seminíferos revestidos únicamente por células de Sertoli, sin presencia de células germinales. Ausencia de signos de malignidad.
- -
Anejo izquierdo: trompa uterina, sin lesiones relevantes. Cintilla gonadal sin presencia de células germinales. Ausencia de signos de malignidad.
Con el diagnóstico definitivo de DGP 46XY, la paciente solicita tratamiento hormonal sustitutivo y reconstrucción genital como varón, por lo que se remite a la Unidad de Trastornos de Identidad de Género de la Comunidad de Madrid.
DiscusiónLa DGP 46XY ha recibido diversos nombres a lo largo del tiempo, desde disgenesia gonadal mixta a seudohermafroditismo masculino disgenético.
La DGP XY se caracteriza por los individuos presentan formación testicular parcial y cariotipo 46XY, genitales externos ambiguos que pueden presentarse casi completamente femeninos o casi masculinos y mezcla de conductos de Wolff y Müller1.
Pueden presentar gónadas en bandeleta (cintillas fibrosas) de un lado, con un teste contralateral disgenético o normal, o ambos testes disgenéticos, o bien un tumor de un lado, con una gónada del otro lado3,4.
Es probable que sea un síndrome genéticamente heterogéneo, por un defecto parcial en el gen del SRY o bien por mutación de otro gen que interactúa con el SRY en estadios tempranos de diferenciación testicular1,5.
La mayoría de los casos son evaluados en el periodo neonatal por genitales ambiguos; 2 de cada 3 son calificados como femeninos: La apariencia de los genitales externos varía; sin embargo, la mayoría presenta un sáculo utrículo-vaginal.
Cuando consultan en la adolescencia, lo hacen por signos de virilización, como hirsutismo o un tono de la voz grave. Un 25% presenta estigmas de síndrome de Turner, con manifestaciones variables (etiología desconocida, probable mosaico no detectado).
La histología gonadal es muy variable. Los testes disgenéticos pueden contener en estadios tempranos células germinales, pero están generalmente ausentes en la adolescencia. En lactantes y niños, el hallazgo más común es la ausencia parcial de la túnica albugínea6.
Los pacientes con disgenesia gonadal que involucra al cromosoma Y tienen mayor probabilidad de presentar tumores de células germinales. Para la DGP 46XY se ha descrito probabilidad de un 25%. El riesgo es bajo en la infancia y se incrementa con la edad, sobre todo después de la pubertad y con mayor incidencia a los 30 años.
La posibilidad es mayor en el testículo disgenético que en la gónada en bandeleta.
Por orden de frecuencia, el gonadoblastoma y el disgerminoma son los tumores que más aparecen. Otros tumores encontrados han sido carcinoma embrionario, tumor del seno endodérmico, teratoma inmaduro y neoplasia intratubular maligna6.
Los genitales externos ambiguos son evaluados generalmente en el nacimiento, siendo el diagnóstico más frecuente en estos casos el de hiperplasia suprarrenal congénita. El caso que presentamos resulta excepcional al tratarse de una persona adulta de 24 años. El diagnóstico de los estados intersexuales en la edad adulta suelen ser consecuencia del estudio de la amenorrea primaria.
Este diagnóstico precisa de un estudio clínico, analítico, genético, radiológico, quirúrgico, anatomopatológico y psicológico, que permita su clasificación exacta y el tratamiento más apropiado, teniendo siempre en cuenta su identidad sexual7.
La clasificación más didáctica recomendada por la SEGO atiende a la estructura de la gónada8, aunque en este caso es el estudio anatomopatológico de las gónadas y el sexo genético los que nos dan el diagnóstico.
Teniendo en cuenta que: a) la definición de gónada disgenética es aquella con ausencia de células germinales; b) los niveles androgénicos no están elevados en este caso para poder etiquetarse como una disgenesia gonadal mixta; c) no existe tejido ovárico completamente desarrollado y sí tejido testicular disgenético, por lo que excluimos el hermafroditismo verdadero, y d) el cariotipo es 46XY, el diagnóstico de este caso es el de DGP XY9-11.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.