


El síndrome de insensibilidad a los andrógenos se caracteriza por la presencia de cariotipo 46,XY, fenotipo femenino y presencia de gónadas masculinas. Es la tercera causa más frecuente de amenorrea primaria, tras la disgenesia gonadal y la ausencia congénita de vagina (síndrome de Mayer-Rokitansky-Küster-Hauser). Se trata de una entidad de interés por su relevancia en la identificación sexual y por su posible asociación con tumores malignos de las gónadas masculinas, que hace necesario un correcto diagnóstico y tratamiento quirúrgico. En este artículo se describen dos casos de síndrome de insensibilidad a los andrógenos, con su estudio clínico-genético y tratamiento, así como su seguimiento.
Androgen insensitivity syndrome is characterized by the presence of external female phenotype, 46,XY karyotype and intraabdominal testes. This syndrome is the third most frequent cause of primary amenorrhea, after gonadal dysgenesis and congenital absence of the vagina (Mayer-Rokitansky-Küster-Hauser syndrome). Androgen insensitivity syndrome is of interest due to its role in sexual identification and its possible association with malignant tumors of the male gonads, which require an accurate diagnosis and surgical treatment. We present two cases of androgen insensitivity syndrome. The results of the clinical and genetic examinations, as well as the treatment and follow-up of these two patients, are discussed.
La insensibilidad a los andrógenos es la tercera causa más frecuente de amenorrea primaria, tras la disgenesia gonadal y la ausencia congénita de vagina. Su frecuencia en la población oscila alrededor de 1/20.000 a 1/60.000 nacimientos1.
Este síndrome fue descrito por primera vez por John Morris2 en 1953, que también lo denominó «síndrome de feminizacion testicular»’, ya que se caracteriza por la presencia de fenotipo femenino asociado a un cariotipo masculino normal 46,XY.
Fue en 1970 cuando Lyon y Hawkes3 revelaron la presencia de un gen ligado al cromosoma X como causa del síndrome de feminización testicular en el ratón. Después de múltiples estudios genéticos, se estableció la localización del gen responsable del receptor intracelular de andrógenos (RA) en el cromosoma X humano4,5. En 1989, se definió la localización exacta de este gen en la región q11-126 del cromosoma X y en el mismo año se demostró que el síndrome de insensibilidad a los andrógenos era causado por mutaciones en este gen7. En 1989 fue cuando se obtuvo el conocimiento completo a nivel molecular del gen RA, determinando su secuencia de intrones y exones8. Aproximadamente, el 70% de las mutaciones en el receptor de los andrógenos están ligadas al cromosoma X y se heredan de manera recesiva. Existen hasta 750 mutaciones del receptor de los andrógenos conocidas que resultan en varias enfermedades que incluyen el síndrome de insensibilidad a los andrógenos completo y el síndrome de insensibilidad a los andrógenos parcial o incompleto (síndrome de Reifenstein)9,10.
Mutaciones en las regiones codificantes y de unión exón/intrón en el gen RA son responsables de impedir el desarrollo de las estructuras derivadas del conducto de Wolff, debido a la falta de inducción ejercida por los andrógenos. Pero a la vez se produce la inhibición de las estructuras derivadas del conducto de Müller por la presencia de la hormona antimulleriana (AMH) sintetizada por las células de Sertoli después de la diferenciación testicular.
El síndrome de insensibilidad a los andrógenos o feminización testicular se caracteriza clínicamente por la presencia de fenotipo femenino, con una vagina corta y ciega (derivada del seno urogenital) y ausencia de útero, trompas de Falopio y ovarios. Las mamas presentan un desarrollo puberal y destaca la ausencia de vello púbico y axilar. Existen testículos que presentan un desarrollo normal, pero habitualmente se encuentran en el abdomen o anillo inguinal. En el perfil hormonal, las concentraciones de testosterona se encuentran dentro de la normalidad o ligeramente elevadas para un varón, la LH elevada, la FSH normal o elevada y el estradiol elevado (para un varón)11.
La presentación típica del síndrome de insensibilidad a los andrógenos es la amenorrea primaria en una adolescente fenotípicamente femenina. En una niña de menor edad, la presentación podría ser en forma de hernia inguinal. Estudios recientes muestran que el 1,1% de las niñas premenárquicas con hernias inguinales presentan el síndrome de insensibilidad a los andrógenos completo, mientras que el 80-90% de las pacientes con síndrome de insensibilidad completo a los andrógenos eventualmente desarrollarán una hernia inguinal12.
En este artículo, presentamos dos casos de insensibilidad completa a los andrógenos recientemente diagnosticados en nuestro servicio.
Casos clínicosCaso 1Paciente de 34 años de edad, natural de Perú, que consulta por amenorrea primaria.
Destaca entre sus antecedentes personales una herniorrafia inguinal bilateral en su país a los 24 años de edad. En sus antecedentes familiares, una tía y prima maternas, también presentan amenorrea primaria, pero no han sido estudiadas.
A la exploración física presenta fenotipo femenino con mamas bien desarrolladas, ausencia de vello axilar y escaso vello púbico. Los genitales externos femeninos son de aspecto normal con una vagina ciega de 6cm. Al tacto bimanual, no se palpan útero ni anejos.
Se practica una ecografía transvaginal que muestra ausencia de útero y ovarios.
Los niveles plasmáticos de 17-B-estradiol son normales (36 pg/ml). Se encuentran valores altos de FSH (21,83 mU/ml), LH (23,88 mU/ml) y testosterona (13,55 ng/ml).
Se realiza un estudio citogenético de sangre periférica con resultado de cariotipo 46,XY.
El estudio genético molecular de los ocho exones del gen RA muestra una mutación puntual missense con cambio de G por A en el codón 787 del exón 6, que predice en la proteína la sustitución del aminoácido metionina por isoleucina (M787I).
Se completa el estudio con una resonancia magnética nuclear pélvica que descarta la presencia de gónadas masculinas y muestra ausencia de anomalías a otros niveles.
Actualmente, la paciente realiza tratamiento hormonal sustitutivo con estrógenos y sigue controles periódicos en nuestras consultas.
Caso 2Paciente de 31 años, natural de Ecuador, que consulta por amenorrea primaria.
Presenta como antecedentes patológicos la exéresis del globo ocular izquierdo a los 12 años de edad, sin destacar otros antecedentes patológicos de interés. En su familia, su hermana y una prima materna también presentan amenorrea primaria.
A la exploración física, presenta fenotipo femenino con mamas bien desarrolladas, ausencia de vello axilar y escaso vello púbico, clitoromegalia leve y labios menores hipoplásicos, con vagina ciega de 7cm. Al tacto bimanual, no se palpan útero ni anejos. A nivel del canal inguinal, de manera bilateral, se palpa una tumoración de 5cm, móvil.
Se practica una ecografía transvaginal que muestra la ausencia de útero y ovarios.
Los niveles plasmáticos de 17-B-estradiol (70 pg/ml) son normales, mientras que se encuentran valores altos de FSH (21,65 mU/ml), LH (28,22 mU/ml) y testosterona (38,64 ng/ml).
Se realiza un estudio citogenético de sangre periférica con resultado de cariotipo 46,XY. El estudio genético molecular muestra una mutación puntual missense en el exón 8 del gen RA, que ocasiona en la proteína la sustitución del aminoácido prolina por serina (Pro913Ser).
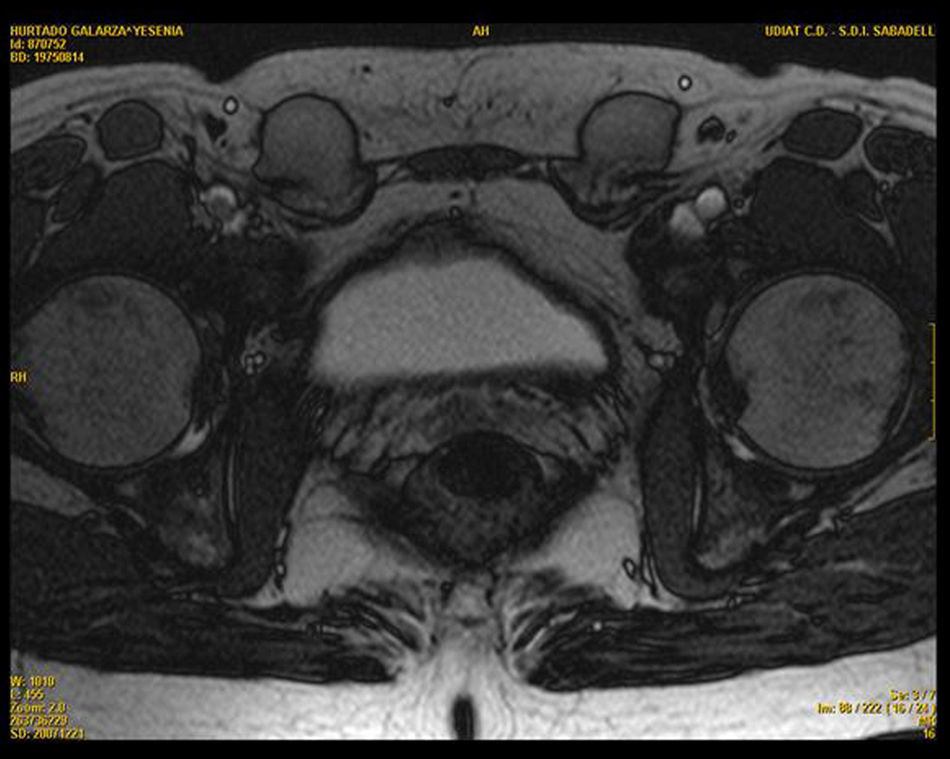
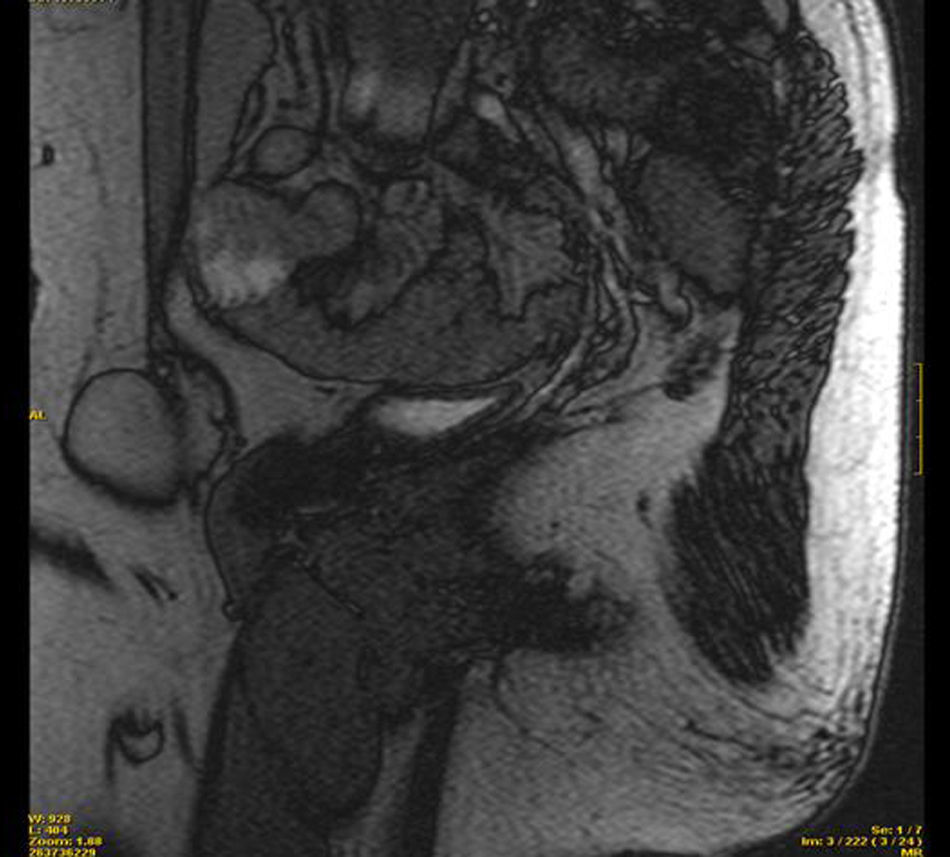
Se completa el estudio con resonancia magnética pélvica que revela la presencia de dos imágenes a nivel del trayecto inguinal bilateral, compatibles con testículos, ambos de morfología normal. Se visualiza una vagina ciega y ausencia de útero y anejos (figs. 1 y 2).


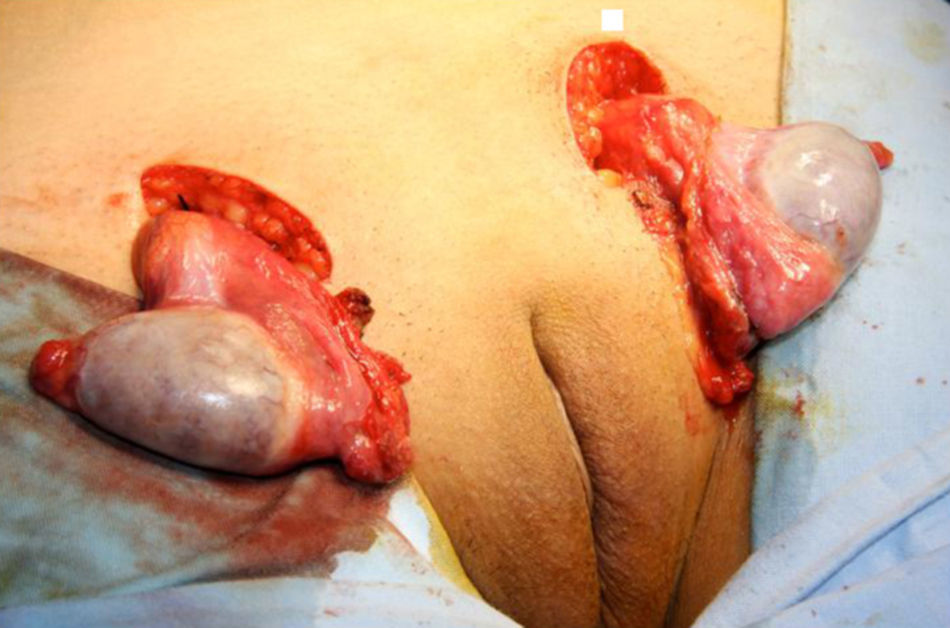
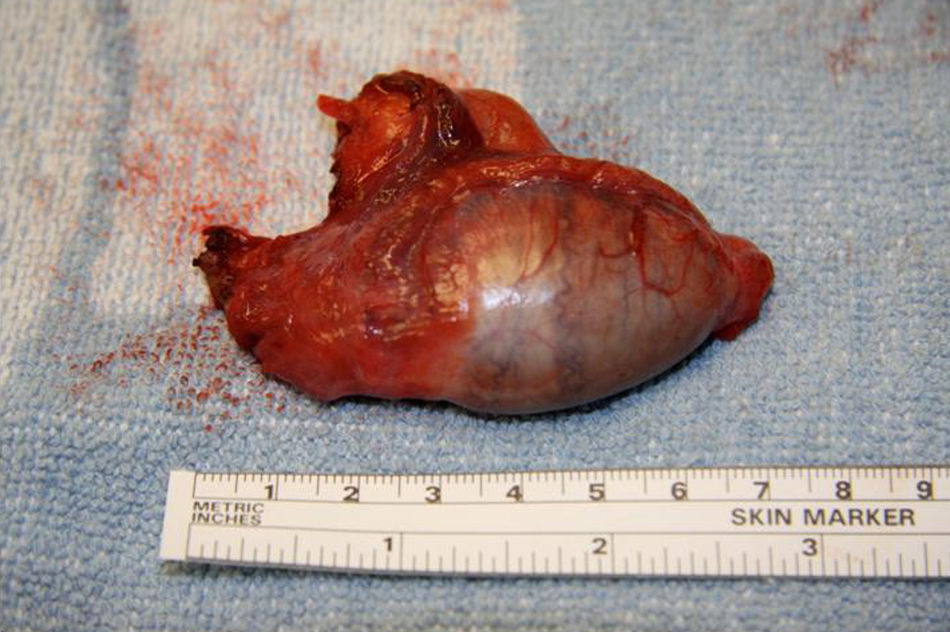
Se practica una gonadectomía quirúrgica laparotómica sin complicaciones. (figs. 3 y 4). El resultado de la anatomía patológica informa de la presencia de parénquima testicular con túbulos seminíferos constituidos por células de Sertoli e intersticio con hiperplasia de células de Leydig.


Actualmente, la paciente se encuentra en tratamiento hormonal sustitutivo.
DiscusiónEl diagnóstico diferencial de la amenorrea primaria incluye la ausencia congénita de vagina (síndrome de Mayer-Rokitansky-Küster-Hauser), la disgenesia gonadal y el síndrome de insensibilidad a los andrógenos. La ausencia congénita de vagina es la causa más frecuente de amenorrea primaria, con una incidencia de 1/5.000. Estas pacientes presentan un desarrollo mamario normal, con una vagina corta, pero normal desarrollo de vello púbico y axilar. El estudio del cariotipo es crucial para el diagnóstico diferencial de la ausencia congénita de vagina y el síndrome de insensibilidad a los andrógenos. Los niveles hormonales de testosterona también nos pueden ayudar en el diagnóstico diferencial, ya que en la ausencia congénita de vagina los niveles de testosterona son normales, mientras que en el síndrome de insensibilidad completo a los andrógenos se encuentran elevados. Podemos distinguir la disgenesia gonadal completa XY (síndrome de Swyer) del síndrome de insensibilidad completo a los andrógenos por la presencia de genitales internos y externos femeninos normales pero infantiles. Existen bandas fibrosas en lugar de gónadas, por lo que existe amenorrea primaria y ausencia de desarrollo sexual secundario al llegar la pubertad. Los estudios de imagen, como la resonancia, nos permiten estudiar la anatomía interna, descartando la presencia de tumores gonadales. Pero el análisis mutacional del gen RA es la prueba definitiva para la confirmación del síndrome de insensibilidad a los andrógenos.
Existen formas incompletas de este síndrome, todas causadas por un trastorno recesivo ligado al cromosoma X. Su frecuencia es 10 veces menor que el síndrome completo. La clínica varía desde la presencia de una feminización casi completa hasta la masculinización fenotípica. Entre ambos extremos existen formas intermedias que se caracterizan por la presencia de clitoromegalia leve y ligera fusión de los labios hasta una ambigüedad genital significativa. En la actualidad, reciben el nombre de síndrome de Reifenstein todas las formas intermedias que inicialmente recibieron nombres individuales. Se han descrito casos de varones en que la única manifestación de insensibilidad a los andrógenos es la presencia de azoospermia u oligospermia intensa. En los casos en que el defecto de la función de los receptores de andrógenos es sutil, pueden existir varones afectados que sean fértiles. El síndrome del varón fértil hipovirilizado es otra manifestación de este trastorno de los receptores de andrógenos.
La presencia de testículos, ya sea intraabdominales o en el trayecto inguinal, resulta importante en el estudio de las pacientes con el síndrome de insensibilidad completo a los andrógenos, ya que existe posibilidad de malignización.
A diferencia de las disgenesias gonadales, en las que los testes intraabdominales deben ser extirpados en el momento del diagnóstico por la alta incidencia de malignización (20-30%), en el síndrome de insensibilidad a los andrógenos estos pueden extirparse después de la pubertad, debido a la aparición relativamente tardía de los tumores gonadales en estos casos y a su menor incidencia (5-10%). Las gónadas masculinas pueden extirparse mediante laparoscopia o laparotomía13–15. Los estudios de imagen, incluida la resonancia pélvica como técnica gold standard, permiten estudiar la anatomía interna y descartar así la presencia de testículos intraabdominales, información necesaria para valorar la necesidad de tratamiento quirúrgico, así como las posibles vías de abordaje del mismo. Si no se dispone de resonancia, el estudio mediante ecografía abdominopélvica puede ser útil.
En estas pacientes, los testes muestran una hiperplasia de las células de Leydig y pueden estar presentes en el canal inguinal (efecto de descenso intraabdominal de la AMH). La existencia de antecedentes de hernias inguinales en la infancia debe hacer sospechar este síndrome.
Las mujeres con síndrome de insensibilidad a los andrógenos completo presentan menor densidad mineral ósea, motivo por el que necesitan tratamientos mantenidos con calcio y vitamina D. También se aconseja la práctica de ejercicio de manera regular.
Es necesario el seguimiento de estas mujeres en consultas especializadas, ya que precisan tratamiento hormonal con estrógenos tras la gonadectomía. Se debe realizar un seguimiento de su densidad mineral ósea mediante densitometría, ajustando el tratamiento en caso de su disminución con bifosfonatos9.
Son importantes el consejo y el asesoramiento psicológico adecuado en estas pacientes, reforzando su identidad sexual femenina, ya que el diagnóstico ocasiona un importante estrés emocional para ellas y su familia. Un psicólogo con experiencia en alteraciones en el desarrollo sexual tendría que formar parte del plan terapéutico lo antes posible; también se recomienda un consejo genético a la familia. Las mujeres portadoras tienen un riesgo del 50% de transmitir la mutación del gen RA en cada uno de los embarazos. Una vez identificada la mutación familiar en el gen RA, se debe ofrecer la posibilidad de estudio molecular a las mujeres para la detección del estado de portadoras. Asimismo es posible un diagnóstico prenatal para los embarazos de mujeres portadoras.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.