





INTRODUCCION
El higroma quístico (HQ) cervical fetal es un defecto congénito, caracterizado por el desarrollo de espacios distendidos y llenos de líquido en la región posterior cervical del feto1.
El origen de esta enfermedad parece ser un problema en el desarrollo del sistema vascular linfático1-10.
Según la literatura médica, se detecta HQ aproximadamente en una de cada 120 ecografías realizadas en el primer trimestre y principios del segundo trimestre, aunque la incidencia al nacimiento es mucho menor (1/6.000-1/12.000 recién nacidos [RN] vivos).
El HQ se asocia a cromosomopatías en el 50-80% de los casos2, así como a diversas anomalías estructurales. Su diagnóstico se acompaña de una alta tasa de procedimientos invasivos. Se asocia con una elevada tasa de abortos espontáneos e inducidos (motivo por el cual su historia natural es poco conocida), así como a una alta mortalidad intrauterina, con una escasa supervivencia libre de enfermedad1,2.
Con todo lo anterior, al diagnosticar un HQ nos planteamos cómo informar a los padres y establecer el tratamiento fetal. Diseñamos este estudio con el objetivo de conocer la incidencia, la evolución y los resultados perinatales del HQ en nuestra población.
MATERIAL Y MÉTODOS
Se trata de un estudio longitudinal, retrospectivo, descriptivo, que incluye los casos de HQ diagnosticados en una muestra de población general no seleccionada, estudiados en la Unidad de Diagnóstico Prenatal (UDP) y Terapia Fetal del Hospital Universitario Materno Infantil de Canarias (HUMIC), entre el 1 de enero de 1992 y el 31 de julio de 2005.
El Hospital Universitario Materno Infantil de Canarias es el único centro de tercer nivel en la isla de Gran Canaria, y el servicio de ginecología y obstetricia es también el único en el ámbito de la asistencia pública. A todas las gestantes se les solicita una ecografía en su primera visita prenatal, que se realiza en la misma UDP, en las consultas externas o en los centros de atención especializada (atendidos por los obstetras del servicio). Los casos de HQ incluidos en este estudio fueron diagnosticados en la UDP o remitidos a esta unidad tras sospecharse la existencia de HQ en una consulta externa, para confirmar el diagnóstico y completar el estudio y el seguimiento. Por tanto, los casos proceden de la población general de gestantes que solicitaron asistencia prenatal en nuestro servicio.
Dado que en los primeros años del estudio (1992-2000) no estaba sistematizada la medición de la translucencia nucal entre las semanas 11 y 14 de gestación, se utilizaba como criterio de realización de técnicas invasivas para el estudio del cariotipo una edad materna superior a 35 años, por lo que en este grupo de población la primera ecografía se hacía coincidir con la fecha de realización de la técnica correspondiente (15 semanas).
Tras la progresiva implantación del cribado combinado del primer trimestre, en los últimos años del estudio ha disminuido la edad gestacional (EG) de realización de la primera ecografía, con diagnósticos más precoces de alteraciones morfológicas.
También se incluyeron los casos diagnosticados tardíamente (después de la semana 18 de EG), que corresponden con pacientes a las que no se les había realizado ecografías previas (por control tardío de la gestación, por error de citación o por inasistencia a consulta), a quienes se diagnosticó un HQ en la ecografía de cribado morfológico del segundo trimestre, que se realiza a toda la población en la UDP entre las semanas 18 y 22 de EG.
Se incluyeron los diagnósticos de HQ en los que se describía un exceso de líquido en la región cervical posterior del feto loculado por uno o más septos.
Las variables analizadas fueron: número de HQ diagnosticados, EG al diagnóstico, edad materna al diagnóstico, número de RN totales y RN vivos, número de RN con diagnóstico prenatal de HQ y su evolución, número de abortos espontáneos en fetos con HQ, número de abortos inducidos tras el diagnóstico de HQ, malformaciones asociadas a HQ, número de pruebas invasivas para el estudio del cariotipo y cromosomopatías asociadas a HQ.
Se seleccionaron los casos de la base de datos de la UDP. Se revisaron sistemáticamente las historias clínicas para comprobar la evolución gestacional. Se comprobaron las anatomías patológicas en los abortos espontáneos e inducidos. Se contactó telefónicamente con las madres de los niños diagnosticados de HQ y nacidos vivos para conocer la evolución posnatal. Los datos referidos a la población general para el cálculo de la incidencia se obtuvieron de la base de datos general del hospital sede del estudio.
RESULTADOS
Durante el período de estudio se diagnosticaron un total de 56 casos de HQ que cumplieran los criterios definidos para la inclusión.
En el período de estudio el número total de RN fue de 102.707, y el número de RN vivos de 101.992.
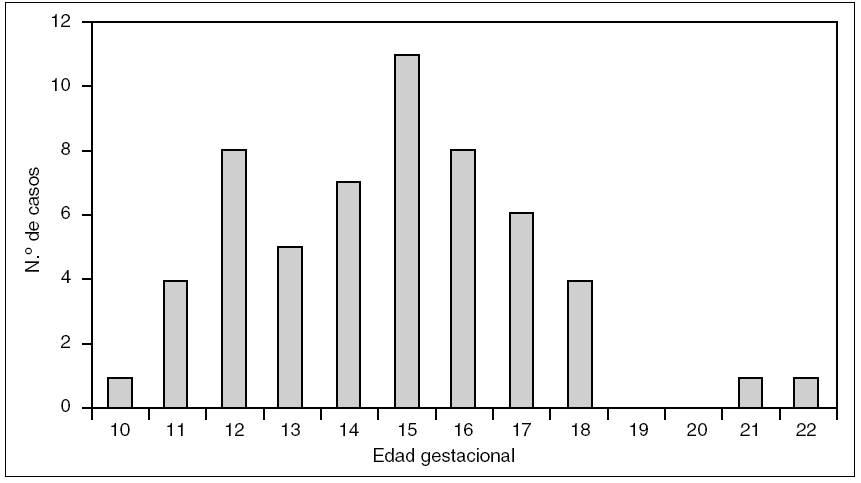
La EG media al diagnóstico fue de 14,6 semanas. La EG con mayor número de diagnósticos fue de 15 semanas. Todos los casos se diagnosticaron antes de las 23 semanas, y el 96% fue diagnosticado antes de las 18 semanas. La distribución de la EG al diagnóstico se refleja en la figura 1.
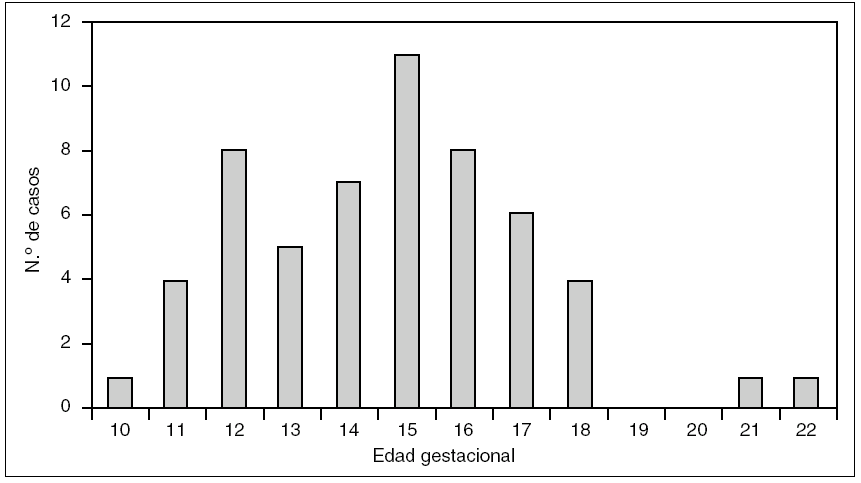
Figura 1.Semanas de gestación al diagnóstico y número de casos de higroma quístico en el Hospital Universitario Materno Infantil de Canarias.
La edad materna (EM) media al diagnóstico fue de 29,53 años. Los 35 años fueron el pico de edad en el que se realizaron más diagnósticos de HQ, aunque el 71% de las pacientes tenía menos de 35 años.
El 70% de los fetos diagnosticados de HQ presentaban anomalías asociadas, y el hidrops fetal (definido como acumulación de líquido en 2 o más cavidades) fue la más frecuente (39%). Las malformaciones asociadas al HQ en nuestra serie se relacionan en la tabla 1.

El 23% (n = 13) de las parejas rehusaron realizar el estudio del cariotipo una vez informadas del diagnóstico. Se realizó un estudio del cariotipo al 77% (n = 43) de los fetos afectados de HQ, y se registraron 2 casos (el 5% de los estudios de cariotipo) sin resultados por falta de crecimiento celular en el cultivo. En la serie presentada, 41 casos de HQ tenían un cariotipo conocido. De ellos, el 63% (n = 26) presentaban un cariotipo anómalo, mientras que sólo el 36% (n = 15) tenía un cariotipo normal, y uno de ellos presentó un polimorfismo (46, XY, 9qh+). En la tabla 2 se incluye la distribución de los cariotipos asociados a HQ en nuestro medio.

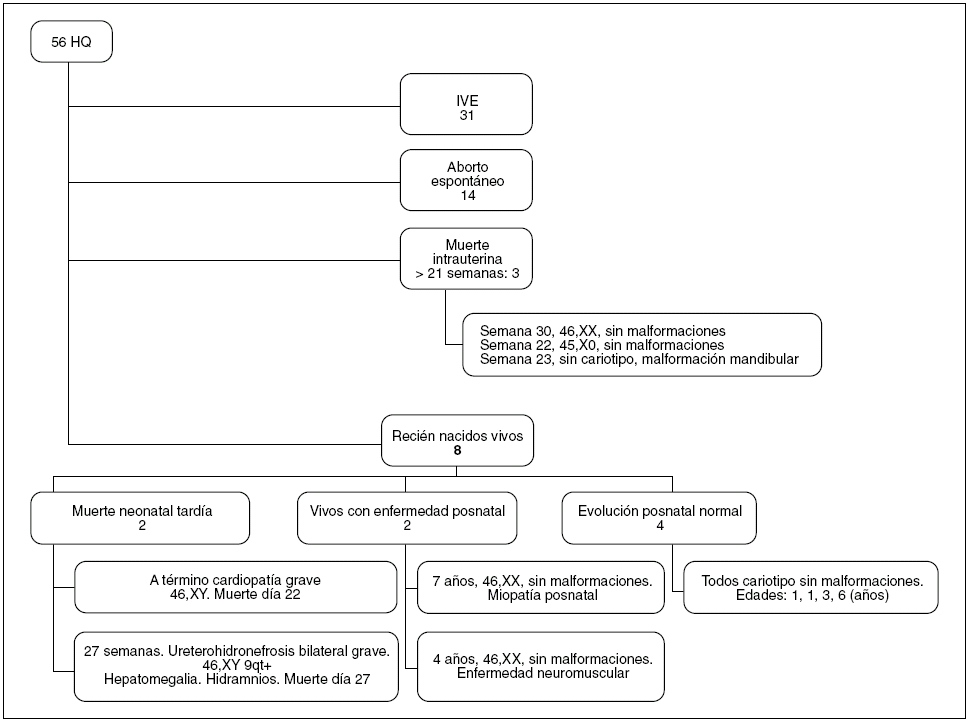
La evolución de los fetos diagnosticados de HQ se refleja en la figura 2. El 55% (31 de 56) de los padres optó por una interrupción voluntaria del embarazo (IVE). De las 25 gestaciones restantes con diagnóstico de HQ que tuvieron una evolución espontánea, 17 terminaron con muerte fetal intrauterina (68%), 14 de ellas antes de la semana 22 y en 3 casos con una EG ≥ 22 semanas. Por tanto, la mortalidad anteparto espontánea fue del 68%. En el subgrupo de fetos con HQ con anomalías asociadas y/o cariotipo anómalo, la mortalidad fue del 100%. Sólo 8 de los 56 fetos diagnosticados de HQ (el 14%) nacieron vivos. De ellos, 2 presentaron una muerte neonatal tardía (ambos tenían HQ, polihidramnios, cariotipo normal y una malformación asociada, una cardiopatía en el primer caso y una hidronefrosis bilateral con hepatomegalia en el segundo caso). Excluyendo las IVE, la mortalidad perinatal fue del 76% (19 de 25 casos) en la serie de HQ. La evolución de los otros 6 RN vivos diagnosticados de HQ se recoge en la tabla 3. La supervivencia total registrada es del 10% (6 de 56 casos), con una supervivencia libre de secuelas del 7% (n = 4), y sólo tuvo lugar en casos de HQ aislado con cariotipo normal.
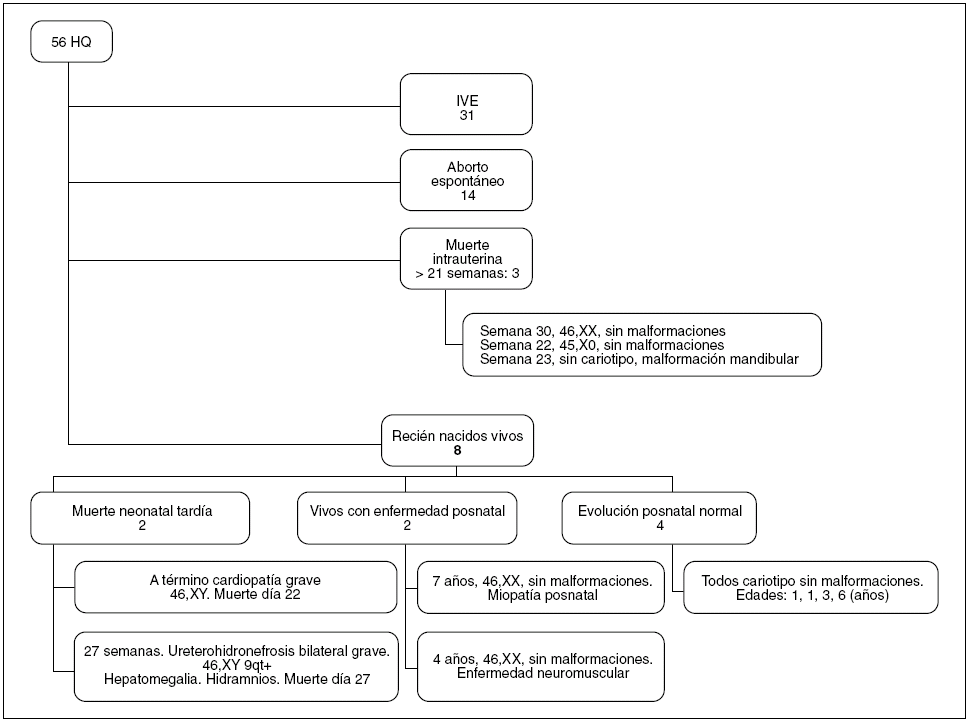
Figura 2. Evolución y resultados perinatales de los casos de higroma quístico (HQ) diagnosticados en el Hospital Universitario Materno Infantil de Canarias. IVE: interrupción voluntaria del embarazo.

La incidencia global de HQ en las gestantes de nuestra población es de 1/1.851 RN vivos.
DISCUSION
La necesidad de ahondar en el conocimiento del HQ y su historia natural, con el fin de asesorar adecuadamente a las parejas a quienes realizamos un diagnóstico de HQ, nos llevó a emprender este estudio con los datos de la población atendida en nuestra UDP.
En nuestro medio hemos observado una incidencia global (total de casos diagnosticados) de HQ de 1/1.851 RN vivos. Sin embargo, dada el alto porcentaje de parejas que optaron por la IVE, y la alta tasa de mortalidad espontánea de esta entidad, sólo uno de cada 12.820 RN vivos (8 RN vivos con HQ/101.992 RN vivos totales) había sido diagnosticado de HQ durante la gestación. Si bien en la literatura médica las cifras de incidencia-prevalencia son muy dispares, podemos decir que nuestros datos se encuadran dentro de lo publicado1-8.
La elevada tasa de aneuploidías en la población de fetos afectados de HQ hace recomendable el ofrecimiento del estudio del cariotipo. Coincidimos con la mayoría de estudios3,5,6,7,9 en que la alteración cromosómica más frecuentemente encontrada en la población de fetos con HQ es la monosomía X, o síndrome de Turner (el 34% de nuestra muestra). No obstante, algunos grupos, como el de Malone1,11 y el de Podobnik8 describen la trisomía 21 como la aneuploidía más frecuente en los HQ (el 37% de trisomía 21 frente al 28,3% de síndrome de Turner, en el grupo de Malone; el 47% de trisomía 21 frente al 11% de síndrome de Turner en el grupo de Podobnik). Desde la introducción en la década de los años noventa del término «translucencia nucal» por el grupo de Nicolaides, para el cribado de aneuploidías del primer trimestre, se ha suscitado un amplio debate en la comunidad científica acerca del significado clínico de la acumulación de líquido en la región cervical fetal. El grupo de Nicolaides defiende en una reciente publicación12 que el higroma quístico no es una entidad distinta de la translucencia nucal incrementada en el primer trimestre, por lo que no se le debe asignar un cociente de riesgo independiente al de la translucencia nucal. Sin embargo, el grupo de Malone1,11 ha publicado varios artículos con las conclusiones del estudio First and Second Trimestre Evaluation of Risk (FASTER), en los que afirma que el HQ (entendido como incremento de translucencia nucal en la longitud total del feto y en el cual hay septos claramente visibles) tiene una probabilidad de aneuploidía fetal del 50%, y debe conducir hacia la realización de una técnica invasiva para el estudio del cariotipo directamente, sin necesidad de asociar al cálculo de riesgo marcadores séricos maternos. Por otro lado, el grupo de Ville9,10 defiende también el HQ como una entidad distinta a la translucencia nucal, y asegura que su diagnóstico prenatal es relevante no sólo por el riesgo de aneuploidía, sino también como una parte importante del consejo genético cuando el cariotipo es normal. Sin embargo, la definición de HQ del grupo francés no coincide con la del grupo de Malone, ya que consideran que el HQ es una malformación congénita del sistema linfático caracterizada por la presencia bilateral de sacos linfáticos yugulares llenos de líquido en la región cervical fetal. Ville coincide con Nicolaides en que el edema subcutáneo cervical o difuso con finos tabiques no es un HQ, sino una forma más del espectro de la translucencia nucal. No obstante, para Ville la importancia de la diferenciación entre HQ y translucencia nucal incrementada radica en los casos con cariotipo normal, ya que en ello el HQ se asocia con mayor frecuencia que la translucencia nucal incrementada a malformaciones fetales que pueden no ser diagnosticadas en estudios ecográficos del primer trimestre, hecho que condiciona la información y el consejo que debe transmitirse a los padres.
La alta incidencia de malformaciones asociadas al HQ hace que, aun con un cariotipo normal, deba realizarse un exhaustivo estudio anatómico fetal2,3,5,7,8. En nuestra muestra, después del hidrops, las cardiopatías son las malformaciones anatómicas más frecuentes. La asociación de malformaciones ensombrece el pronóstico del HQ, con una mortalidad del 100%.
A la vista de los resultados del estudio, podemos concluir que el HQ es una enfermedad con mal pronóstico, con una elevada mortalidad (en torno al 76%) y una supervivencia libre de secuelas muy baja (el 7% de nuestra muestra), que sólo tiene lugar en los HQ aislados y con cariotipo normal.
De acuerdo con los grupos de Malone1 y Tanriverdi7, proponemos la siguiente secuencia ante el diagnóstico de un HQ:
1. Información general acerca del diagnóstico y el pronóstico del HQ. Ofertar el estudio del cariotipo en todos los casos.
2. a) Si el cariotipo es anómalo, informar de la posibilidad de IVE. b) Si prosigue la gestación y el cariotipo es normal, proponer un estudio morfológico, sobre todo para descartar el hidrops y las cardiopatías.
3. a) Si se identifican malformaciones asociadas, valorar su pronóstico e informar a los padres. Valorar IVE. b) Si el cariotipo es normal y no hay malformaciones asociadas, informar adecuadamente del pronóstico a los padres, y que éstos decidan en consecuencia.
Correspondencia:
Dra. E. Cortés Cros.
Servicio de Ginecología y Obstetricia. Hospital Universitario Materno Infantil de Canarias.
Avda. Marítima del Sur, s/n. 35016 Las Palmas de Gran Canaria. España.
Correo electrónico: ecortescros@yahoo.com
Fecha de recepción: 17/1/2006.
Aceptado para su publicación: 9/2/2007.