






INTRODUCCION
Las malformaciones congénitas de la mama son un proceso muy frecuente; sin embargo, casos como el que presentamos son extremadamente raros: se ha encontrado sólo uno en la literatura médica, publicado en 1994 por Berman y Davis1. Este caso tiene la particularidad de presentar tejido mamario ectópico en las axilas que secreta directamente a la piel, sin presentar pezón supernumerario. Sí es más frecuente, y así lo demuestra la literatura médica, la secreción láctea por la axila con un pezón supernumerario o dependiente de la cola de la mama2.
CASO CLINICO
Se trata de una paciente de 33 años, con 2 gestaciones y 2 partos previos, hace 6 meses y 2 años, respectivamente, en ambos casos con lactancia materna posterior. Tras el primer parto la lactancia se desarrolló con normalidad, pero 8 días después del segundo parto la paciente acudió a urgencias por secreción por ambas axilas de forma espontánea.
En la exploración en decúbito supino, con las manos detrás de la cabeza, presentaba secreción por ambos pezones a la presión, y secreción espontánea a través de varios puntos de la piel de ambas axilas. No se halló ningún pezón supernumerario en las 2 líneas paraesternales ni en las axilas, si bien al tacto en ambas axilas se apreciaba una zona con bordes mal definidos, con aumento de la densidad, y ligeramente dolorosa. Macroscópicamente la secreción axilar presentaba las mismas características que la de los pezones. Posteriormente, se realizaron un estudio ecográfico de las mamas y axilas, una citología de la secreción de las axilas y una ecografía renal.
Ecografía de la mama. Las mamas eran de aspecto normal. Había tejido mamario en las dos axilas; en la izquierda, había una colección líquida con aspecto de galactóforo dilatado y en la derecha, un quiste simple de 4 x 2 mm (figs. 1-3).
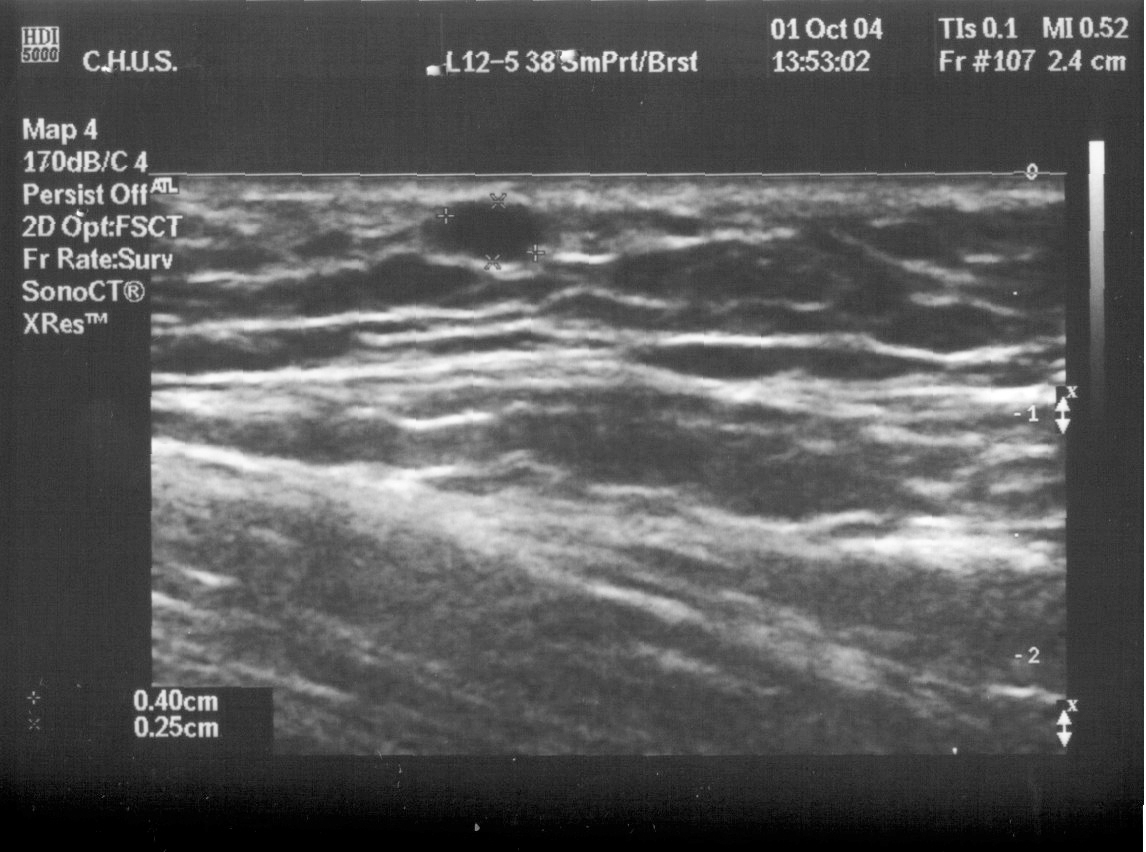
Figura 1.Quiste axilar.
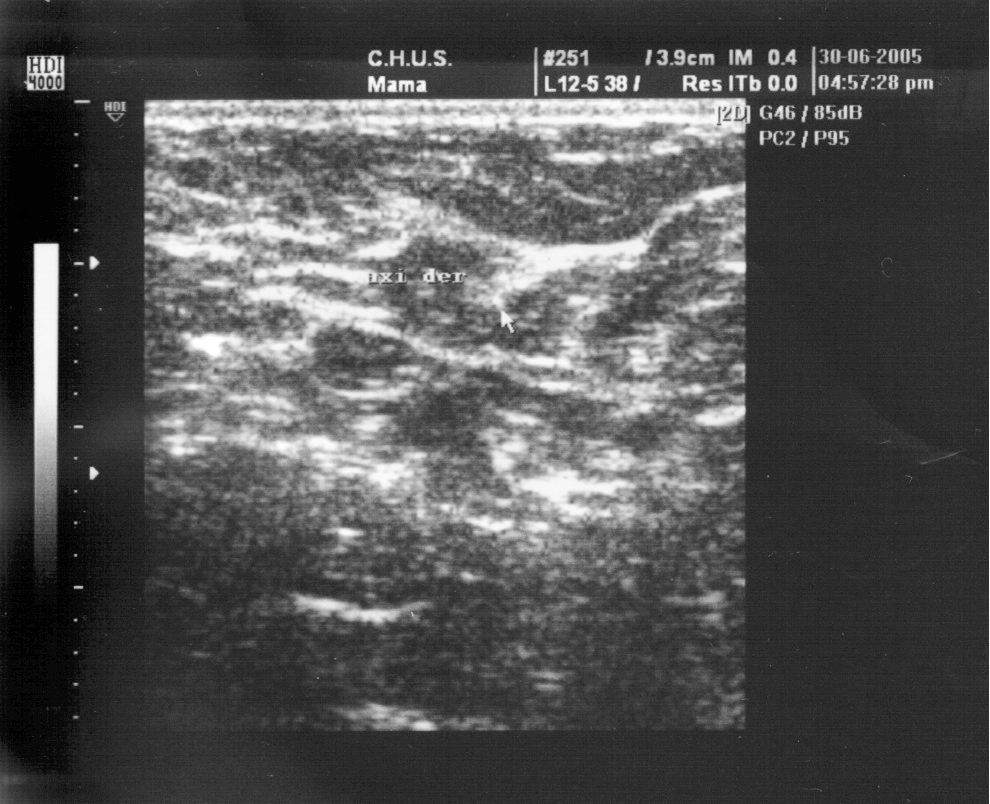
Figura 2.Axila derecha al año del parto.
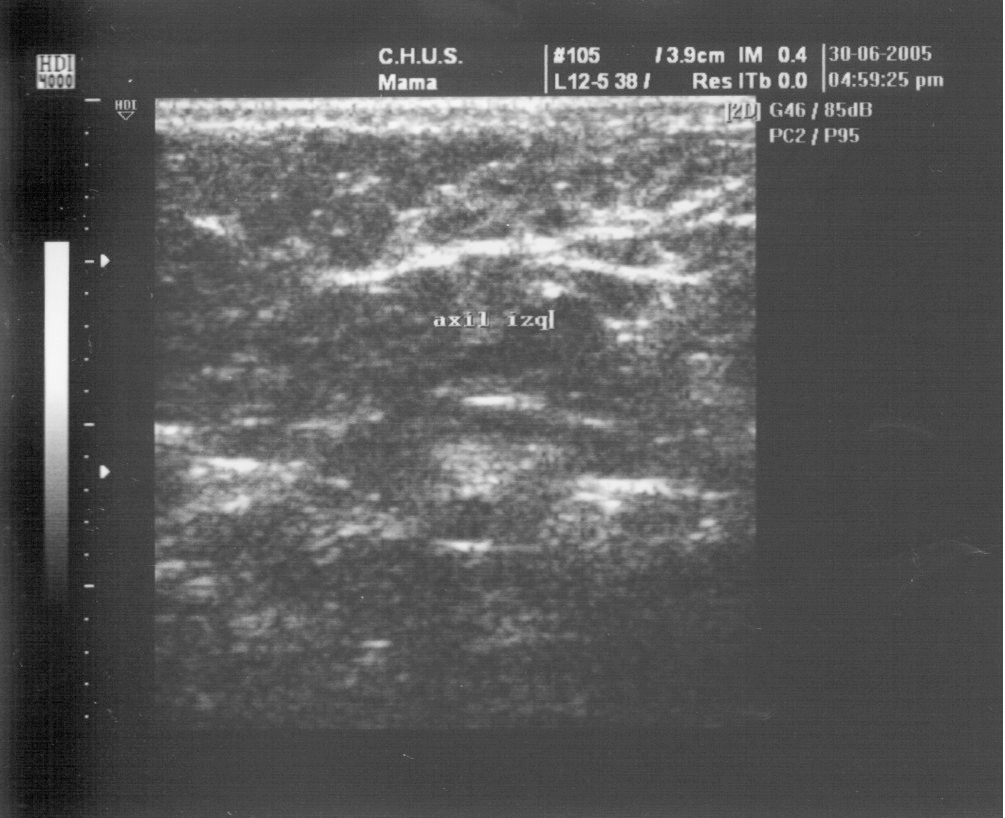
Figura 3.Axial izquierda al año del parto.
Anatomía patológica. Frotis con fondo proteináceo sobre el que se apreciaron células de características macrofágicas de citoplasma espumoso y núcleos redondos, uniformes y normocromáticos. No se observaron células atípicas; era compatible con secreción láctea.
Ecografía renal. Ambos riñones estaban en la localización habitual y presentaban una ecogenicidad normal.
Completado el estudio clínico y de pruebas complementarias, se llegó al diagnóstico de secreción láctea por ambas axilas a través de la piel, sin pezón supernumerario.
La paciente continuó con lactancia materna y se realizó una nueva revisión a los 8 meses tras el parto. En ese momento sólo daba una toma diaria y en la exploración la secreción por las axilas sólo se producía con la presión, ya no de forma espontánea, y sólo por un punto de la piel. El estudio ecográfico mostró tejido mamario en las axilas, pero ya no formaciones quísticas.
Al año del parto la paciente dejó la lactancia y en un nuevo estudio ecográfico se encontró tejido mamario normal. No presentaba secreción espontánea ni con la expresión.
DISCUSION
La existencia de las malformaciones congénitas de la mama está en su embriología. Ésta es similar a la de otras glándulas de la piel; así, su origen parece estar en el ectodermo3, del que deriva la epidermis. En la cuarta semana de gestación se puede observar en el embrión 2 líneas ventrales, paralelas a la línea media, que son lo que los anglosajones llaman milk lines; en estas líneas se encuentran las células que originarán las glándulas mamarias y se disponen desde la axila hasta los labios mayores. En los seres humanos desaparecen, excepto en la cuarta línea intercostal, donde comienza a desarrollarse la mama definitiva. A partir de la quinta semana de desarrollo embrionario se produce un divertículo de la epidermis que se hunde en la dermis (originada del mesodermo). Este divertículo empieza a crecer y en la décima semana se ramifica; al final de la gestación llega a tener de 15 a 20 lóbulos. Las células de estos nuevos canalículos entran en contacto con varias hormonas durante el desarrollo fetal; como la progesterona, la hormona de crecimiento, los estrógenos o la prolactina, sobre todo en la semana veinte de gestación4.
Las malformaciones congénitas de la mama son frecuentes y pueden aparecer hasta en un 10% de la población, según algunas series5. Se pueden clasificar de muy distintas maneras, pero una forma lógica y tradicional sería en función de la existencia o no de tejido mamario supernumerario.
Malformaciones con tejido mamario supernumerario
Ocurre en el 2-6% de las mujeres6 y en el 1-3% de los varones5. Existen, a su vez, muchas subclasificaciones de este grupo. Una de las más clásicas es la de Kajava de 1915 (tabla 1)7.

Nuestro caso sería una clase IV de Kajava, ya que no tenía pezón ni aréola, pero sí tejido glandular. El tipo más común es la clase VI o politelia; su localización, en un 90% de los casos, es inframamario. La mayoría de las veces no se diagnostica pues el paciente lo confunde con un nevo y no consulta por él. Tradicionalmente se ha relacionado con la politelia con la presencia de malformaciones urológicas; hoy día existen trabajos que afirman que esta relación es estadísticamente significativa8. Así, la presencia de quistes renales, las duplicaciones o la agenesia de uno de los 2 riñones aparece en un 1-2% de la población general frente a un 14- 32% de los pacientes con politelia (dependiendo de si la politelia es esporádica o familiar)5.
La polimastia sería de la clase I a la IV; la última es la más común. Su frecuencia es mayor en mujeres; aparece hasta en un 1-2% de la población femenina. Su localización más común es la axilar5. La mayoría de las veces la polimastia se pone de manifiesto con los cambios hormonales, ya sea la menstruación, el embarazo o la lactancia.
Malformaciones con ausencia total o parcial de tejido mamario
Este grupo es mucho menos frecuente que el anterior; en estos casos puede ser unilateral o bilateral. La situación más grave es la amastia, que sería la ausencia total de tejido glandular, sin pezón y sin aréola. La aplasia sería también la ausencia de tejido mamario, pero con pezón y aréola, presentación clásica del síndrome de Poland, que se acompaña de otras deformidades osteomusculares. Un grado menos grave que estos dos sería la hipoplasia, que se encuentra en el síndrome de Turner.
El diagnóstico, una vez conocidos los tipos de malformaciones congénitas de la mama, en la mayoría de los casos es de a simple vista. Aunque en algunos pacientes puede ser necesario el uso de exploraciones complementarias como la ecografía, ante la sospecha de una polimastia; la mamografía, también usada en la polimastia para hacer un cribado de cáncer de mama en otras localizaciones9, y estudios urológicos complementarios, como la ecografía renal, para el cribado de malformaciones renoureterales.
En cuanto al abordaje terapéutico de estas malformaciones, es muy variable y existen en la literatura médica opiniones contrapuestas sobre qué hacer6,10-12. Todos coinciden en que muchas de estas malformaciones pueden llegar a suponer, fundamentalmente, consecuencias psicológicas y estéticas en quien las presenta. Algunos autores proponen que ante una polimastia axilar como la de nuestro caso clínico sería adecuada la exéresis del tejido mamario accesorio. Sin embargo, existen trabajos en los que se afirma que estas exéresis, en su mayor parte basadas en la estética, conllevan un aumento significativo de la morbilidad.
Existen varios artículos en la literatura científica que hacen referencia a la presencia de tejido ectópico en la axila1,2,6,9,10-12, pero sólo encontramos un caso en el que además haya secreción axilar, que es lo que también ocurre en esta paciente. Ese otro caso al que nos referimos es el de Berman y Davis1, publicado en 1994. Los 2 son muy similares: en ambos el tejido ectópico se pone en evidencia en el momento de la lactancia, cuando aparece secreción en la axila sin pezón accesorio. La única diferencia es que la paciente del caso de Berman y Davis1 presenta anomalías urológicas y nuestra paciente no. Es, por tanto, un caso excepcional por su rareza, en el que la labor médica queda restringida al hecho de conocer su existencia y tranquilizar a la paciente.
Correspondencia:
Dr. M.E. Pérez-Muñuzuri.
Xosé Pasín, 3, 4.o, B. 15706 Santiago de Compostela. A Coruña. España.
Correo electrónico: elena_munuzuri@hotmail.com
Fecha de recepción: 9/6/2006.
Aceptado para su publicación: 15/9/2006.