


El síndrome de Meckel es un infrecuente y letal síndrome polimalformativo de herencia autosómica recesiva. Su incidencia oscila entre 1:1.300 y 1:140.000 nacimientos. Las malformaciones que se observan con más frecuencia son a nivel del sistema nervioso central (frecuentemente meningoencefalocele occipital), displasia renal multiquística bilateral, fibrosis hepática y polidactilia. La alteración genómica que lo produce puede localizarse en diversos loci. El diagnóstico prenatal es posible mediante sospecha ecográfica y, en muchos casos, mediante estudio del ADN fetal. Presentamos el caso de una paciente que tuvo 2 embarazos consecutivos en los que se diagnosticó dicho síndrome.
Meckel syndrome is an infrequent and lethal syndrome characterized by multiple malformations. Inheritance is autosomal recessive. The incidence of this syndrome ranges from 1:1,300 to 1:140,000 births. The most common malformations are those affecting the central nervous system (usually occipital meningoencephalocele), bilateral renal cystic dysplasia, fibrotic changes of the liver and polydactyly. The associated genomic alteration can involve distinct loci. Prenatal diagnosis is feasible with ultrasound findings and, in numerous cases, with fetal DNA analysis. We present the case of a woman who had two consecutive pregnancies with a diagnosis of Meckel syndrome.
El síndrome de Meckel-Gruber fue descrito inicialmente en 1822 por Meckel1, que publicó el caso de 2 recién nacidos gemelos con encefalocele occipital, polidactilia, fisura palatina y riñones multiquísticos. En 1934 Gruber2 describió varios casos de recién nacidos no viables con encefalocele, displasia multiquística renal y frecuentes polidactilias, ambigüedad genital y fibrosis hepática y pancreática. En 1969 Opitz y Howe3 definieron el «síndrome de Meckel» y sus características clínicas. Posteriormente se han descrito casos en diferentes lugares del mundo y se han descubierto las alteraciones genéticas que lo causan4.
Su incidencia oscila entre 1:1.300 (en los indios gujarati) y 1:140.000 nacidos vivos. Se observan diferencias importantes entre las diversas poblaciones, con elevada incidencia en la población belga y la finlandesa. Se transmite mediante herencia autosómica recesiva5,6.
Se trata de una enfermedad letal en la mayoría de los casos, dada la gravedad de las malformaciones que la conforman. Las anomalías que se observan con más frecuencia son a nivel del sistema nervioso central (muy frecuentemente meningoencefalocele occipital), displasia renal multiquística bilateral, fibrosis hepática (casi en la totalidad de los casos) y polidactilia (presente en el 80% de los casos).
Se han descrito numerosas malformaciones dentro de este síndrome. Los criterios diagnósticos aún no han sido completamente definidos, si bien al menos 2 de las 3 malformaciones clásicas (meningoencefalocele occipital/otras anomalías del sistema nervioso central, displasia renal multiquística y polidactilia postaxial) son fundamentales para llegar al mismo y están presentes en la mayoría de casos7,8.
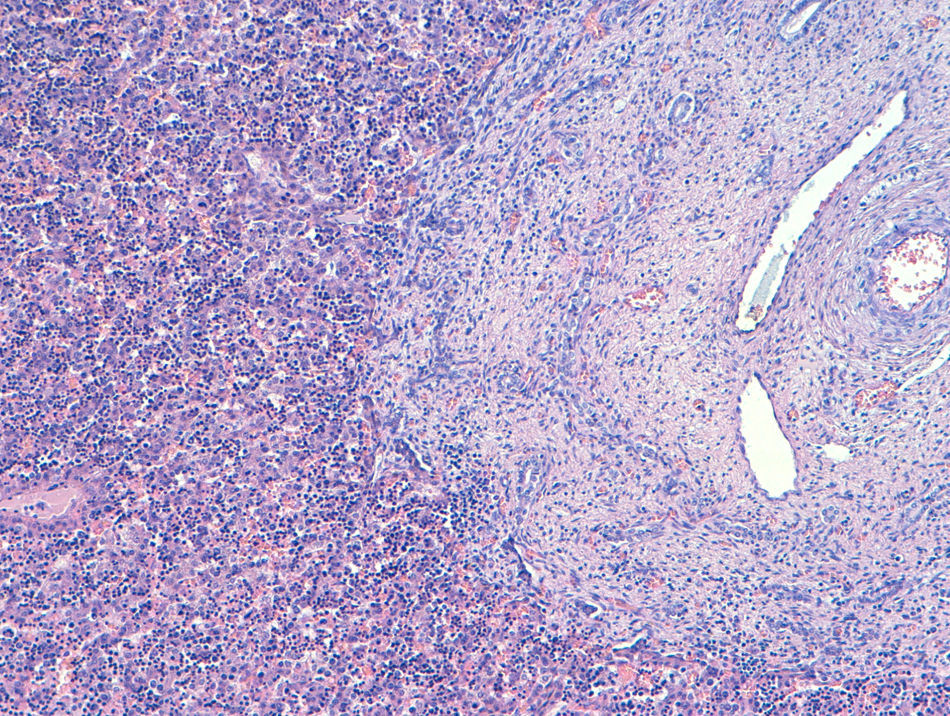
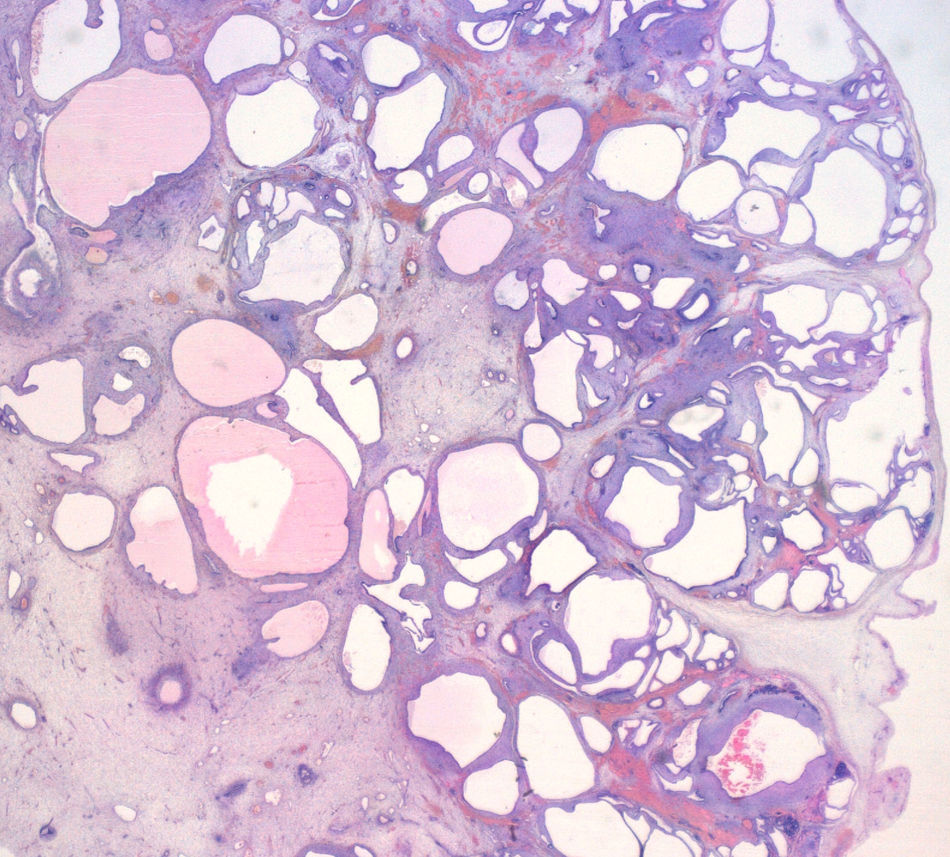
Caso clínicoPaciente árabe de 32 años en 2009, cuando fue diagnosticada en consulta obstétrica de un aborto diferido de 15 semanas de gestación (sg). Se trataba de su cuarto embarazo, tras un parto normal y 2 cesáreas (una en su país, de indicación desconocida, y otra en nuestro hospital en 2007, por presentación podálica y trabajo de parto prematuro de la que nació un feto con ictiosis lamelar que falleció a los 4 meses de vida). No refería otros antecedentes de interés. Su pareja era un varón sano de 30 años consanguíneo (primo). Tras aplicar 3 comprimidos de misoprostol vaginal (Cytotec®), según protocolo empleado en nuestro servicio, se produjo la expulsión del feto y se realizó un legrado obstétrico. El estudio anatomopatológico mostró un feto varón acorde a 15sg, con un síndrome polimalformativo múltiple compatible con un síndrome de Meckel, que presentaba un meningoencefalocele a través de fontanela posterior, fibrosis portal difusa (fig. 1), displasia quística difusa renal bilateral (fig. 2), aesplenia y onfalocele que contenía asas intestinales. Los genitales fueron normales. La placenta no mostró anomalías.
 en el primer feto.")

Tres meses tras el legrado le fue realizada una revisión ginecológica completa que fue normal. No se planteado estudio o asesoramiento genético.
En 2010 volvió a consulta de obstetricia con un nuevo embarazo en el que la paciente desestimó técnicas de diagnóstico prenatal debido a sus creencias religiosas. La ecografía del primer trimestre fue normal a las 12+6sg, con un pliegue nucal de 2,5mm, hueso nasal visualizado y ningún marcador ecográfico de cromosomopatía. La paciente no acudió de nuevo a consulta hasta la semana 22+5 (y por tanto no se realizó la ecografía de nivel II en torno a la 20sg recomendada por la Sociedad Española de Ginecología y Obstetricia), momento en el que refirió una rotura inmadura de membranas de un día de evolución. En la ecografía realizada se observó anhidramnios y un feto vivo con biometría acorde a 18+3sg en el que se apreciaba alguna malformación craneal (poco definible debido a la dificultosa valoración secundaria al anamnios) y una posible displasia renal. Tras explicar a la paciente el inevitable mal pronóstico, se le ofreció una interrupción del embarazo, pero la gestante decidió continuar el mismo, por lo que fue ingresada durante 4 días en reposo, con antibioterapia de amplio espectro (amoxicilina/ácido clavulánico 1g/8h i.v. y gentamicina 240mg/24h i.v.), con control de constantes cada 8h y con control analítico cada 48h. Posteriormente fue dada de alta con indicación de reposo y de amoxicilina/ácido clavulánico 1g/8h vía oral, tras 48h sin mojar y sin mostrar signos de infección durante el ingreso.
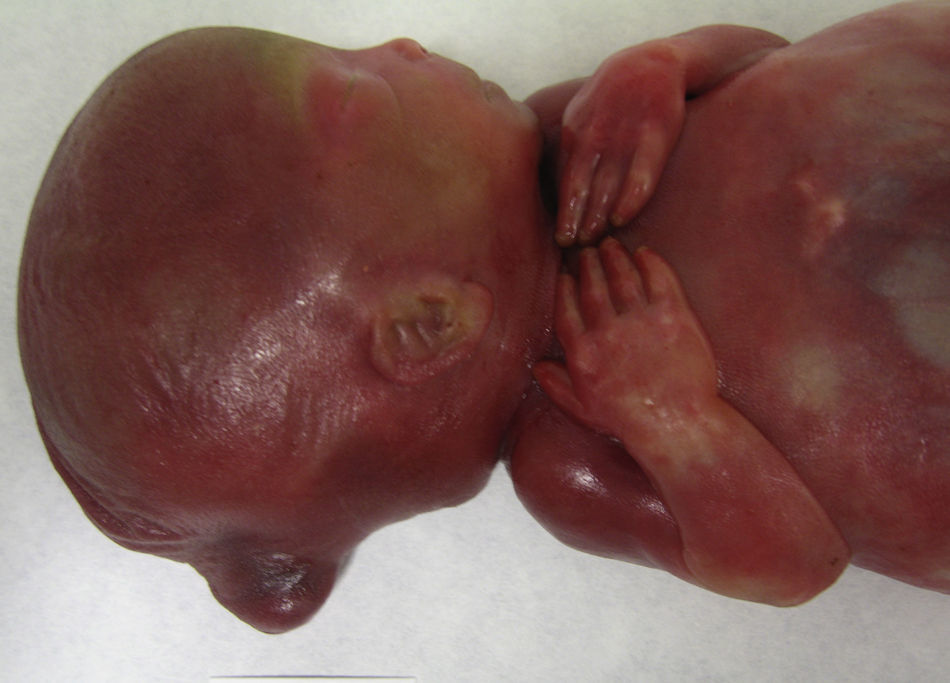
La paciente consultó por urgencias a los 9 días del alta hospitalaria por dinámica uterina subjetiva. A la exploración no presentaba modificaciones cervicales, pero se decidió su ingreso en observación. A las 24h se observó leucocitosis con desviación izquierda (24.500 leucocitos/μl, 91% neutrófilos), fiebre de 38,4°C y ningún foco infeccioso aparente. Ante la sospecha de corioamnionitis, se explicó el mal pronóstico a la paciente y se decidió finalizar la gestación, para lo que se aplicaron 3 comprimidos de misoprostol vaginales (Cytotec®) y posterior estímulo con oxitocina i.v. (cuando la paciente ya tenía 3cm de dilatación cervical). Finalmente se produjo un parto del feto ya sin vida en presentación podálica y un alumbramiento espontáneo, tras el cual se realizó un legrado uterino con legra roma. La observación macroscópica del feto en el momento del nacimiento mostraba un defecto de cierre de pared craneal posterior y orejas de implantación baja.
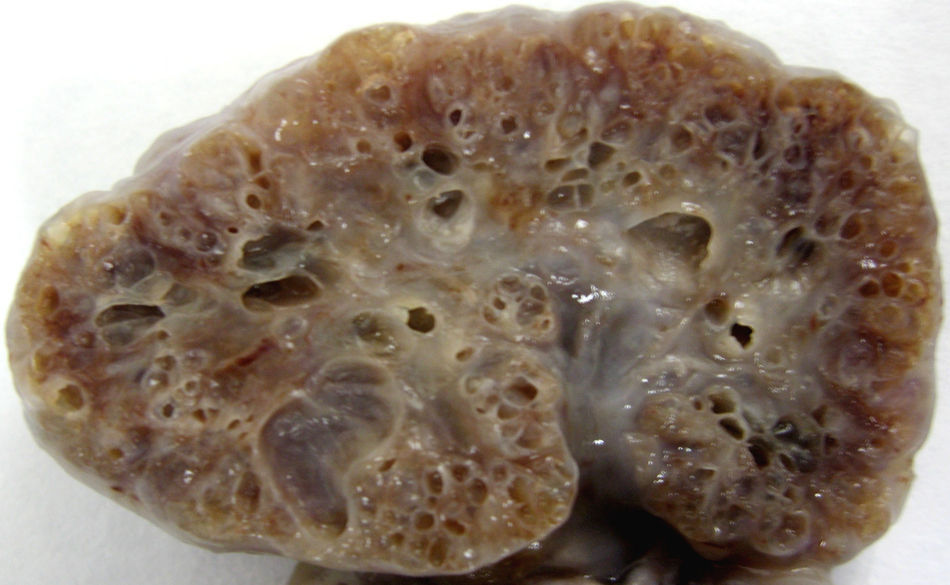
El estudio anatomopatológico macroscópico mostró un feto de 616g con genitales ambiguos (sin lograr localizar testículos), con una protrusión en la región occipital cefálica (fig. 3), pabellones auriculares de implantación baja y riñones grandes (6×5×4cm cada uno) de superficie abollonada y aspecto quístico (fig. 4). El análisis microscópico mostró un meningoencefalocele occipital, displasia quística difusa renal bilateral y fibrosis periductal hepática moderada. No se observaron malformaciones en el resto de órganos. Los hallazgos fueron así compatibles con un síndrome de Meckel.


La paciente fue informada del diagnóstico y remitida a la Unidad de Genética del área sanitaria, donde se realizó un cariotipo a ambos progenitores que fue normal. Posteriormente no se ha podido realizar un análisis genético debido a la situación familiar de la pareja (creencias religiosas y estancia discontinua en España), a pesar de explicarles detalladamente la situación.
DiscusiónPresentamos el caso de una paciente con 2 embarazos consecutivos cuyos fetos cumplen con los criterios diagnósticos típicos del síndrome de Meckel. En el primero de los fetos diagnosticados se objetivó, en el estudio anatomopatológico, meningoencefalocele a través de fontanela posterior, fibrosis portal difusa, displasia quística difusa renal bilateral, aesplenia y onfalocele. En el segundo feto también se llegó al diagnóstico anatomopatológico de síndrome de Meckel, tras observar la presencia de meningoencefalocele occipital, displasia quística difusa renal bilateral, fibrosis hepática periductal y genitales ambiguos. Es llamativo encontrar en 2 ocasiones y en la misma paciente este raro síndrome polimalformativo, si bien es conocido que se transmite mediante herencia autosómica recesiva.
El diagnóstico del síndrome de Meckel se fundamenta en la presencia de al menos 2 de las 3 malformaciones clásicas: meningoencefalocele occipital/otras anomalías del sistema nervioso central, displasia renal multiquística y polidactilia postaxial. Otra malformación constante es la fibrosis hepática. Estos hallazgos son así considerados como criterios mayores para el diagnóstico7,8. Los 2 casos descritos presentaron 3 criterios diagnósticos mayores cada uno.
Existen otras múltiples malformaciones que han sido descritas dentro de este síndrome, si bien su frecuencia es menor. Por ejemplo, fisura palatina (45% de casos), malformaciones cardíacas (20%), de la lengua, a nivel esplénico (como en el primero de los casos descritos), oculares (microftalmia, displasia retiniana, catarata congénita, etc.), faciales (migrognatia, orejas de implantación baja, boca ancha y labios engrosados, etc.), malformación de Dandy-Walker, onfalocele (como también se observó en el primero de nuestros casos), anomalías genitales (como en el segundo caso descrito), etc.6,9.
Las malformaciones a nivel del sistema nervioso central son muy variables; por ejemplo, defectos del cuerpo calloso y de la línea media en general, ausencia de bulbos olfatorios, ausencia de ventrículos laterales, hidrocefalia, etc. La más frecuente (aparece en el 90% de los casos) es la microcefalia con meningoencefalocele occipital, que habitualmente protruye a través de un defecto del hueso occipital o bien de una fontanela posterior alargada. En ocasiones se puede observar un defecto óseo a nivel occipital basal. También son frecuentes las disgenesias de la fosa posterior (quistes en fosa posterior, agenesia de vérmix, etc.)6,9. En ambos casos descritos se observó la presencia de meningocele, la anomalía más frecuente.
La displasia renal bilateral también es muy frecuente (prácticamente constante), con riñones de gran tamaño ya observables en el segundo trimestre y multiquísticos (si bien la histología de su parénquima es normal). Los quistes de las áreas superficiales suelen ser de pequeño tamaño, mientras que conforme se profundiza en la estructura renal su tamaño es mayor. Además afectan a toda la nefrona, mientras que en la displasia renal multiquística —patología también autosómica recesiva— se localizan fundamentalmente en los conductos colectores y son todos pequeños (1-2mm)6,8,10-12. Ambos casos descritos cumplieron también con este criterio diagnóstico.
También en la práctica totalidad de los casos se observan fibrosis hepática y engrosamiento y dilatación de los conductos biliares, típicamente más llamativas en el área portal4,6,8. Los casos descritos de nuevo cumplieron con este criterio diagnóstico.
La aparición de fibrosis en otros órganos ha sido también descrita frecuentemente, por ejemplo en páncreas, bazo, pulmones o ganglios linfáticos. En general, la proliferación de tejido conectivo y la dilatación de los conductos epiteliales asociados en los distintos parénquimas son un hallazgo típico del síndrome de Meckel6.
La polidactilia postaxial bilateral en manos y pies aparece en el 80-90% de los casos. En gestaciones a término es típico encontrar las piernas flexionadas alrededor de un abdomen muy aumentado de tamaño. Los huesos largos pueden aparecer algo acortados y curvados, si bien no existe displasia ósea real6,7. Los casos descritos fueron así infrecuentes, pues no se objetivaron anomalías en las extremidades en ninguno de ellos. Como ya se ha comentado, este es un criterio mayor pero no indispensable para el diagnóstico.
Respecto al área genital, es típico el desarrollo anormal de los genitales masculinos de fetos 46XY. En ocasiones incluso se observa un seudohermafroditismo masculino con presencia de testículos intraabdominales y órganos genitales internos más o menos desarrollados; también se ha descrito la presencia de un hermafroditismo verdadero, con existencia de testículo y ovarios simultáneamente6-9. En el primero de los casos expuestos la edad gestacional y la calidad de las muestras no permitieron valorar detalladamente el área genital. En el segundo de los casos se observó la presencia de ambigüedad genital, sin lograr la localización de testículos extra ni intraabdominalmente.
El diagnóstico diferencial debe plantearse, en primer lugar, con cromosomopatías como la trisomía 13 (síndrome de Patau) o la trisomía 18 (síndrome de Edwards). En estos casos, la realización de un cariotipo sería diagnóstica. El síndrome de Patau asocia con frecuencia malformaciones del sistema nervioso central (p. ej., holoprosencefalia, agenesia de cuerpo calloso, mielomeningocele, etc.), riñones poliquísticos en el 31% de casos, polidactilia en manos y a veces en pies, anomalías oculares y cardiovasculares, pero nunca fibrosis hepática. En el caso del síndrome de Edwards, los hallazgos clínicos son también diferenciadores: microcefalia y holoprosencefalia (no encefalocele posterior), hipotelorismo, fisura palatina y labio leporino típicos (no muy frecuentes en el síndrome de Meckel), polidactilia en manos pero muy infrecuente en pies (típicamente afecta a manos y pies en el síndrome de Meckel), displasia pancreática (la fibrosis hepática es infrecuente) y frecuentes anomalías cardíacas y oculares4,9.
En segundo lugar, se debe plantear el diagnóstico diferencial con otros síndromes polimalformativos, centrado fundamentalmente en el estudio ecográfico prenatal de las malformaciones presentes y, en su caso, en la valoración anatomopatológica y/o genética del producto de la concepción. Así, por ejemplo, es necesario establecer el diagnóstico diferencial con el síndrome de Smith-Lemli-Opitz, un síndrome polimalformativo autosómico recesivo con posibles anomalías del sistema nervioso central (pueden incluir hipoplasia o ausencia del cuerpo calloso, holoprosencefalia, etc.), faciales, genitourinarias y renales, microcefalia, polidactilia y alteraciones hepáticas, así como otras malformaciones diversas, causado por un defecto enzimático en la síntesis del colesterol. En estos casos, la presencia de una frente amplia, ptosis bilateral, pliegues en epicanto y pliegues palmares transversales es definitoria. El diagnóstico se basa en la detección de niveles reducidos de 7-dehidrocolesterol y es posible el estudio de las mutaciones causantes9. En el caso del síndrome de Bardet-Biedl, se trata de un cuadro de herencia autosómica recesiva en el que es típico observar polidactilia, displasia renal en la región medular (aunque no siempre), hipogonadismo y anomalías hepáticas, pero no encefalocele13. También es preciso completar el diagnóstico diferencial con el síndrome de Beemer-Langer (que asocia típicamente polidactilia y costillas cortas y en ocasiones también displasia renal), el síndrome de Joubert (de transmisión autosómica recesiva pero genéticamente heterogéneo, caracterizado por una malformación congénita en el tronco cerebral y una agenesia o hipoplasia del vermis cerebeloso y que asocia con frecuencia displasia renal y polidactilia), el síndrome de Senior-Loken (cuadro autosómico recesivo caracterizado por displasia renal, ocular y a veces hepática que se manifiesta típicamente durante el primer año de vida, pero que no asocia polidactilia ni encefalocele), el síndrome de Jarcho-Levin o disostosis espondilocostal autosómica recesiva (caracterizada por anomalías vertebrales asociadas de manera variable con anomalías del sistema nervioso central como meningocele, tracto genitourinario, displasia renal y corazón) y otros cuadros polimalformativos infrecuentes como el síndrome de Salonen-Herva-Norio. En los casos de enfermedad renal poliquística, tanto dominante como recesiva, es infrecuente la aparición de otras malformaciones asociadas, lo cual es fundamental para la orientación diagnóstica4,12,14,15.
Por último, también es necesario un diagnóstico diferencial con otras asociaciones malformativas, como por ejemplo con los casos de Dandy-Walker, que además presentan riñones multiquísticos. Esta diferenciación es difícil y muchas veces requiere un análisis genético6.
Con respecto a su etiología, el síndrome de Meckel es una enfermedad autosómica recesiva en cuya patogénesis parece estar implicado más de un locus, si bien en algunas áreas se conoce la afectación más frecuente. Así, en Finlandia el locus alterado es el 17q23, en el gen MKS1 (que codifica un polipéptido relacionado con la función ciliar)16,17, mientras que en población del norte de África se observan anomalías en el locus 11q13 (gen MKS2)18; otro estudio las ha encontrado en el 8q21.13-q22.1, en el gen MKS3 (que codifica una proteína transmembrana)19. Estas alteraciones se traducirían en la fibrosis y la formación de quistes en diversos órganos. La existencia de diversas posibles mutaciones causantes del síndrome de Meckel hace necesario conocer la mutación causal de un «caso índice» para poder establecer el diagnóstico genético en otros casos.
La sospecha diagnóstica prenatal es fundamentalmente ecográfica, si bien en ocasiones puede detectarse una elevación de alfa-fetoproteína en sangre materna6. Ya en las 11-12sg podría ser diagnosticado, especialmente en familias con riesgo elevado, mediante la visualización de anomalías en el sistema nervioso central, riñones agrandados e incluso polidactilia20,21. Desafortunadamente en el caso presentado no se observó ninguna anomalía en la ecografía del primer trimestre. En edades gestacionales más avanzadas la displasia renal, el anhidramnios, las malformaciones del sistema nervioso central y el gran perímetro abdominal facilitan el diagnóstico, si bien en ocasiones la ausencia de líquido amniótico puede dificultar la observación de que los riñones ocupan toda la cavidad abdominal4,8. En el caso descrito tampoco fue posible el diagnóstico ecográfico, ya que, tras la visita de primer trimestre, la paciente no volvió a consulta hasta que presentó una rotura de membranas, momento en el cual el anhidramnios hizo imposible precisarlo.
En todos los casos es necesario un detallado examen anatomopatológico para llegar al diagnóstico.
En los casos de familias con mutación conocida, el diagnóstico genético prenatal es posible6. De nuevo el caso descrito es poco afortunado, ya que no es posible realizar dicho estudio y, por tanto, establecer un diagnóstico genético prenatal en próximas gestaciones. El diagnóstico de este síndrome hace además obligatorio un adecuado asesoramiento pregestacional a la pareja ante nuevos embarazos.
El pronóstico es siempre fatal, con una mortalidad del 100% (la mayoría de casos mueren intraútero o a las pocas horas de vida). La confirmación diagnóstica mediante autopsia fetal es fundamental para valorar el riesgo de recurrencia en sucesivas gestaciones4,6,8.
A pesar de las limitaciones encontradas en el diagnóstico y seguimiento del caso expuesto, su presentación tiene por objetivo recordar este síndrome infrecuente pero letal e insistir en la trascendencia del diagnóstico ecográfico y, en su caso, genético.
ConclusiónEl síndrome de Meckel es un infrecuente síndrome polimalformativo letal de herencia autosómica recesiva. Su diagnóstico prenatal es posible mediante sospecha ecográfica y, en muchos casos, mediante estudio del ADN fetal. La confirmación diagnóstica mediante autopsia fetal es fundamental para valorar el riesgo de recurrencia en sucesivas gestaciones.
FinanciaciónPara le realización de este trabajo no se ha contado con ningún soporte financiero.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.