


El síndrome de microcefalia-coriorretinopatía-linfedema es una enfermedad rara con expresividad variable y distintos tipos de herencia. El pronóstico desde el punto de vista neurológico también es variable. Presentamos un caso de esta patología dentro del diagnóstico diferencial de la microcefalia intraútero.
El diagnóstico intraútero de microcefalia es complicado debido a la difícil valoración del desarrollo neurológico que presentarán estos fetos al nacimiento, por lo que el asesoramiento a los padres debe realizarse con sumo cuidado utilizando todos los métodos y algoritmos diagnósticos a nuestro alcance.
Microcephaly-lymphedema-chorioretinal dysplasia is a rare disease with variable expression and distinct forms of inheritance. The neurological prognosis also varies. We report a case of this syndrome as an entity within the differential diagnosis of intrauterine microcephaly. Intrauterine diagnosis of microcephaly is complicated by the difficulty of predicting the degree of neurological development reached by these fetuses at birth. Consequently, great care should be taken when providing parental counseling, using all the diagnostic methods and algorithms available.
El síndrome de microcefalia-coriorretinopatía-linfedema es un cuadro muy poco frecuente descrito por primera vez como asociación por Feingold et al1 y Bartoshesky y Limwongse2. Este cuadro se caracteriza por presentar microcefalia, linfedema de miembros superiores e inferiores, displasia coriorretiniana y distintos grados de retraso mental. Se trata de una entidad monogénica con una expresividad variable cuya herencia puede ser autosómica dominante3, recesiva4 e incluso casos esporádicos5–7.
El primer caso de este síndrome fue publicado en España por Martínez Ruiz7. Presentamos, según nuestro conocimiento, el segundo caso publicado en España de la enfermedad como una entidad dentro del diagnóstico diferencial de la microcefalia intraútero.
En el ámbito mundial, la literatura es escasa y está formada por pequeñas series de casos como las de Gupta et al8, Vasudevan et al9 o Trzupek et al10.
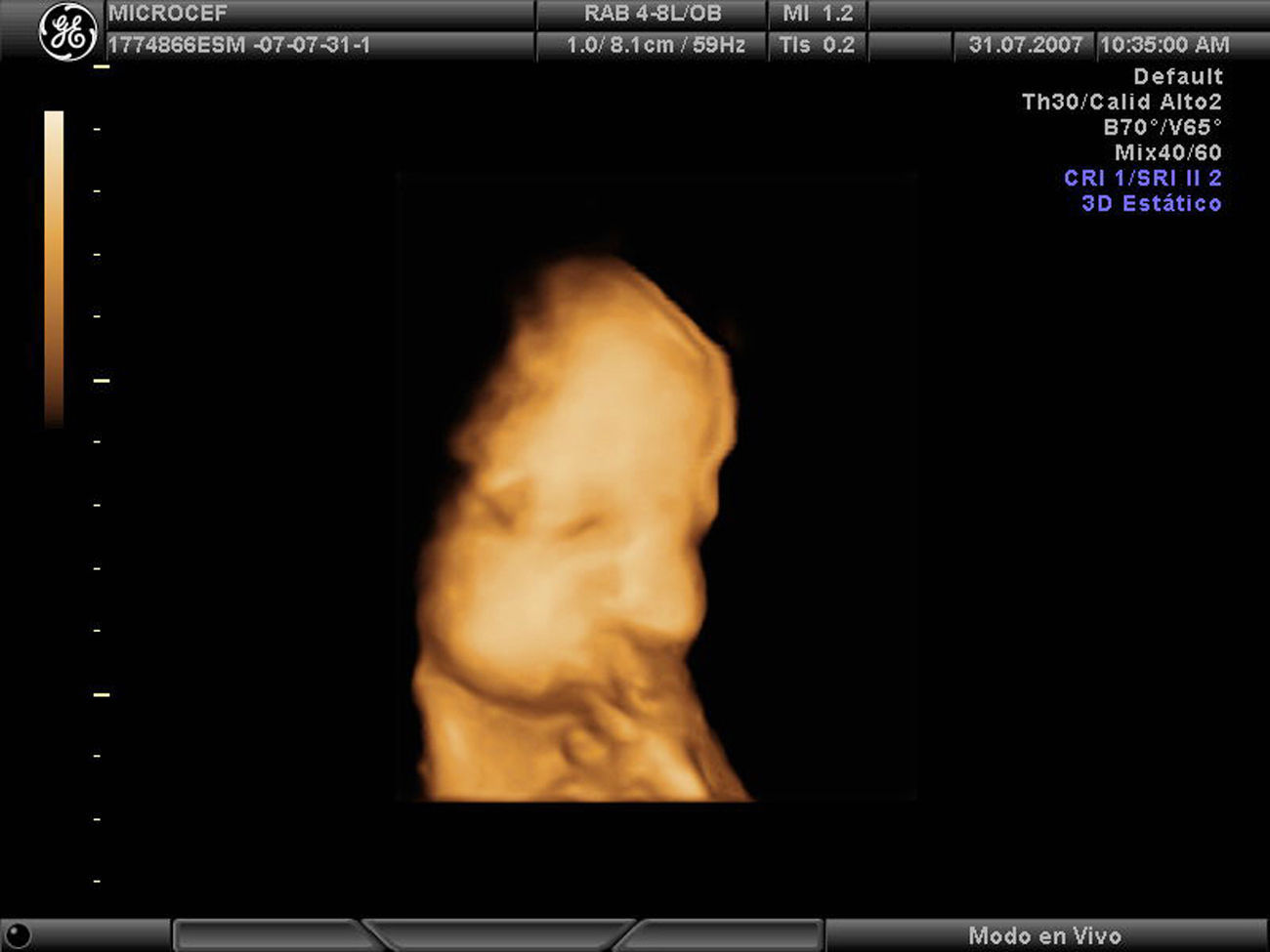
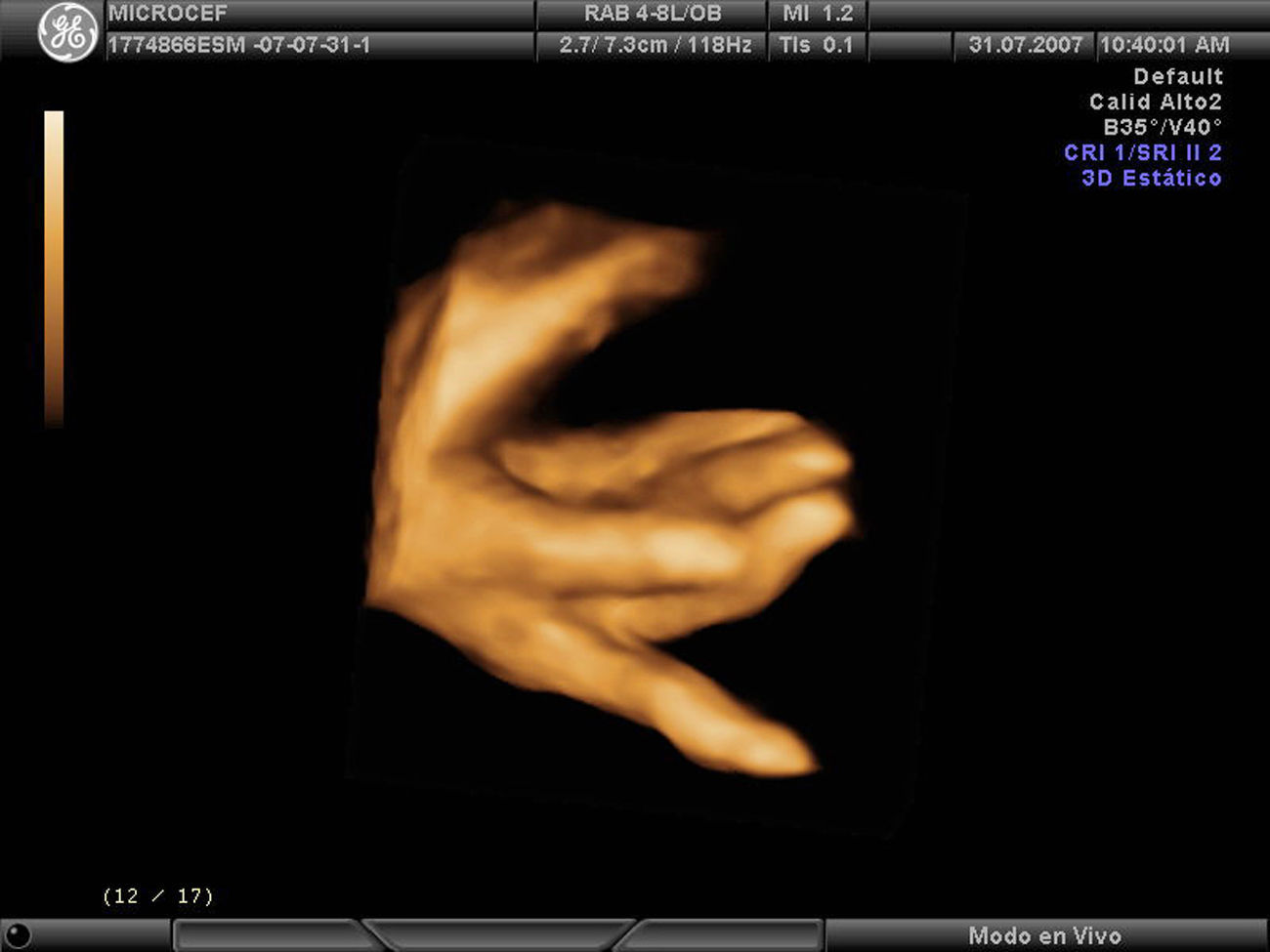
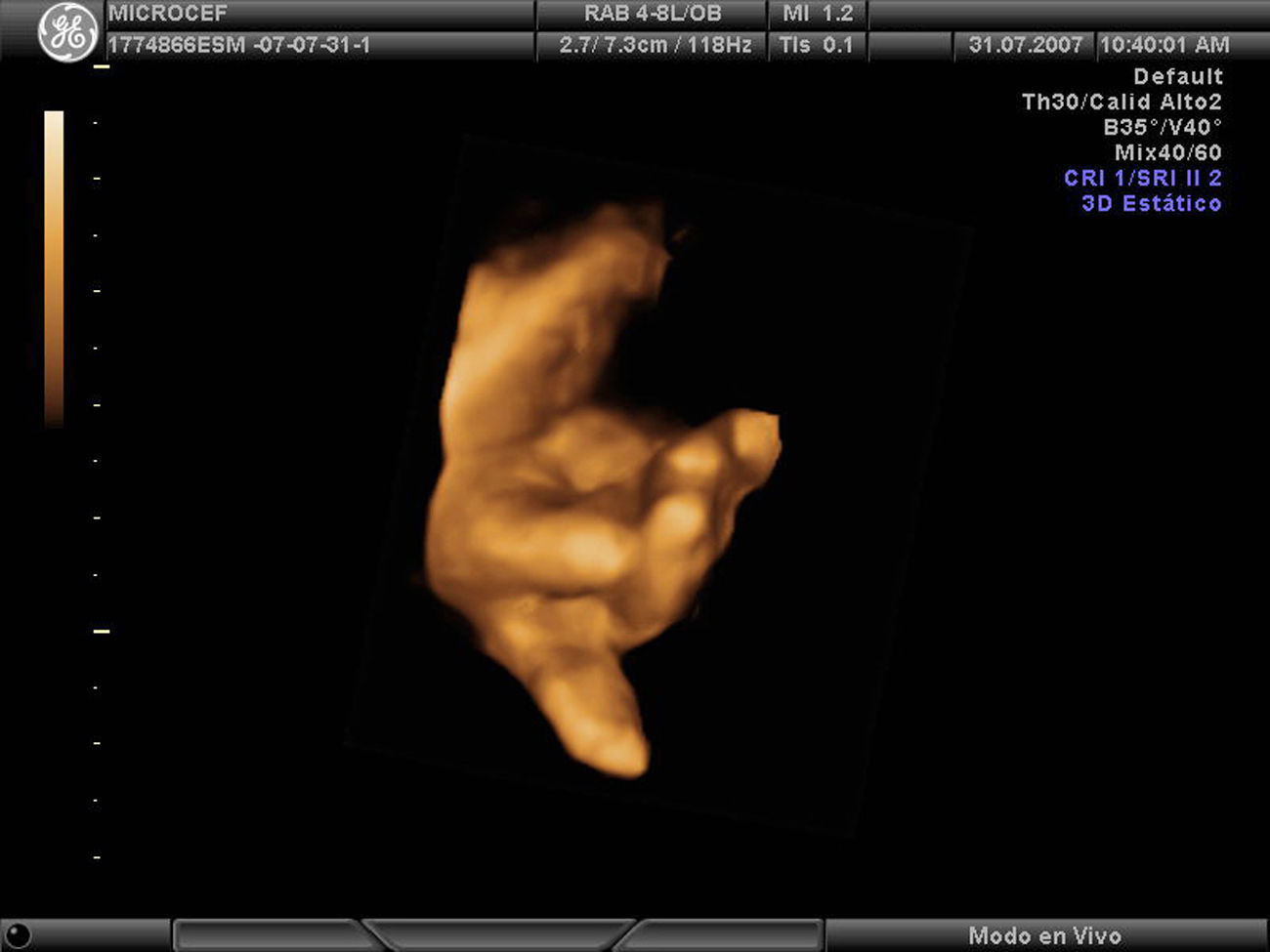
Caso clínicoPaciente de 28 años, secundigesta, fumadora, sin antecedentes personales ni familiares de interés. Remitida a nuestro servicio en la semana 19 para la ecografía del segundo trimestre. Dicha ecografía muestra una biometría unas 2 semanas menor, por lo que se cita nuevamente en 15 días para tratar de descartar un error en la datación. En la ecografía de la semana 21 continúa dicho desfase, con un DBP acorde a 18 semanas y dificultad para evaluar correctamente las estructuras encefálicas por lo que se sospecha un CIR precoz y se cita nueva ecografía en 2 semanas. Se repite el estudio en la semana 24, con hallazgos similares: biometría de 21 semanas y DBP de 18 semanas, sin que se objetiven defectos estructurales. Se presenta el caso en el comité de perinatología del hospital y se decide realizar pruebas para determinar la causa del CIR precoz con microcefalia. Se descartan, en primer lugar, las causas infecciosas tras obtener una batería de serologías maternas para TORCH que resultan negativas, y para descartar causas debidas a alteraciones cromosómicas, se propone funiculocentesis. También se realiza ecocardiografía que resulta normal. En la ecografía de la semana 27 nos ponemos en comunicación con el servicio de neurología infantil para evaluación conjunta del caso. Continúa existiendo un desfase de 3 semanas, DBP y perímetro cefálico 6 semanas menor (fig. 1) y el índice de resistencia en arteria umbilical comienza a estar ligeramente aumentado (IPAU 1,68) siendo el IPACM normal. No se encuentran alteraciones en suturas, corteza, ventrículos cerebrales ni núcleos de la base. La fosa posterior es aparentemente normal y presenta ligeras alteraciones faciales, como micrognatia. La funiculocentesis se realiza a la semana 29 y resulta un cariotipo 46XX normal. En la comisión de perinatología del servicio se continúa debatiendo el caso y se barajan otras causan que también se excluyen, como el consumo de alcohol, fármacos o antecedentes de alguna enfermedad metabólica. Se investigan también los antecedentes familiares de microcefalia, sólo parece existir un tío materno con microcefalia pero con una inteligencia normal. En la semana 31 el desfase es de 5 semanas con DBP 8 semanas menor, en la semana 33 de 6 semanas con DBP 9 semanas menor y, en la semana 35, de 5 semanas con DBP 9 semanas menor, líquido amniótico ligeramente disminuido y resistencia vascular aumentada en arteria umbilical (IPAU 1,8). El estudio anatómico fetal muestra microcefalia posiblemente por craneosinostosis, mala visualización de la fosa posterior, cristalinos opacificados y mano derecha en flexión forzada (figs. 2 y 3).



Con el diagnóstico de CIR precoz con microcefalia marcada (DBP por debajo de 3 desviaciones estándar para EG) y con un cuadro sobreañadido de insuficiencia placentaria con inicio de redistribución vascular, se decide terminar la gestación en semana 35 de común acuerdo con la paciente. Debido a la inmadurez cervical, se realiza una cesárea. Nace una mujer que no se pesa con Apgar 8/9, pH de cordón 7,27 y reanimación tipo II, que se traslada a la REA.
El estudio posnatal muestra un peso de 1.300g (p<25), talla 41cm (p<10) y perímetro cefálico 25cm (p<10). Se evidencia, además, microcefalia marcada sin craneosinostosis, microrretrognatia, linfedema marcado en los pies que afecta a la planta, el dorso y los dedos, hipertonía de miembros superiores e inferiores, no se consigue orientación visual y pobre atención al entorno. Se realizó cribado de infecciones (varicela-zóster, virus herpes simple I y II, citomegalovirus, virus de Epstein-Barr, virus herpes 6) que resultó ser negativo.
El estudio de pruebas complementarias muestra en la resonancia magnética un patrón de giros simplificados y posible hipoplasia discreta del vermis cerebeloso, y la ecografía transfontanelar presenta un cuerpo calloso fino. En el estudio de fondo de ojo se aprecia una coriorretinopatía y aplasia papilar en ambos globos oculares. Dichas pruebas se complementaron con la realización de un estudio de fondo de ojo de ambos progenitores que resulto ser normal.
A la vista de los hallazgos se diagnóstica al recién nacido de síndrome de microcefalia-linfedema-coriorretinopatía, aunque la microcefalia es más marcada de lo habitual para dicho síndrome, con clínica de disfunción neurológica.
DiscusiónLa microcefalia consiste en una cabeza desproporcionadamente pequeña respecto a la edad gestacional y el tamaño del cuerpo. La tasa de incidencia al nacimiento es de 1/6.250 hasta 1/8.50011. Como ya hemos comentado, este síndrome se caracteriza por microcefalia, linfedema en miembros inferiores y displasia retiniana, aunque también se han descritos defectos faciales como retrognatia o implantación baja de las orejas y defectos cardíacos como alteraciones septales1,2,5–10,12–15.
Ecográficamente el diagnóstico de microcefalia debe sospecharse si el DBP es menor de 3 desviaciones estándar que la media para la edad gestacional. El diagnóstico de microcefalia intraútero no siempre se correlaciona con los hallazgos posnatales, de hecho existe un estudio donde sólo se confirmó el diagnóstico tras el parto en 4 de 24 fetos con medidas por debajo de lo normal11.
Generalmente, el diagnóstico de microcefalia implica un fallo en el desarrollo del cerebro, cuanto menor es la circunferencia de la cabeza menor es el nivel intelectual, salvo en el caso de las craneosinostosis donde la inteligencia se encuentra respetada11. Nuestro caso presenta desde el inicio una marcada microcefalia, que se confirma tras el parto, y que hace suponer que el desarrollo intelectual de este recién nacido va a estar comprometido.
Existen múltiples causas prenatales de microcefalia como factores genéticos, ambientales, asfixia, infecciones (típicamente por CMV), fenilcetonuria materna, fármacos, alcohol y radiaciones5,6. Puede acompañarse de alteraciones en las estructuras encefálicas, como ventriculomegalia, esquizencefalia y alteraciones de la inducción ventral16.
Nuestro caso presenta una importante microcefalia desde la semana 21-24 (fig. 1) que se confirma tras el parto, donde la circunferencia cefálica está por debajo del percentil 10. La evaluación prenatal de las estructuras encefálicas junto con el neurólogo pediátrico no presenta alteraciones llamativas aunque destaca la dificultad para la exploración de la fosa posterior. En nuestro caso fuimos planteándonos diferentes causas que progresivamente se iban descartando. Se barajaron: infecciones, que se descartaron mediante serologías maternas; alteraciones cromosómicas, descartadas mediante la realización de un cariotipo fetal a través de funiculocentesis; la paciente no presentaba antecedentes de ninguna enfermedad metabólica ni consumo de fármacos o alcohol. Asimismo en la última exploración ecográfica aparecen otras alteraciones anatómicas acompañantes como la opacificación de los cristalinos y la mano en flexión forzada (fig. 2), que posnatalmente se correspondía con una hipertonía de los miembros.
El diagnóstico del síndrome se realizó posnatalmente a la vista de los resultados de las pruebas oftalmológicas y la visualización del linfedema al nacimiento. Parece tratarse de un cuadro esporádico ya que no existen el la familia antecedentes de dicho síndrome, solo un tío materno parece tener microcefalia pero con inteligencia normal. A diferencia de lo publicado, aquí si parecen existir ciertas alteraciones en las pruebas de imagen cerebral y cierta clínica neurológica.
La falta de información que tenemos de esta entidad patológica nos plantea una importante dificultad diagnóstica antenatal. La microcefalia diagnosticada intraútero es un cuadro grave, porque cuanto mayor sea, más riesgo de compromiso en el desarrollo intelectual tendrá el recién nacido y, por tanto, debemos transmitírselo de ese modo a los padres. La prudencia en casos menos evidentes que el aquí presentado es fundamental al dar el pronóstico a los padres, porque muchas microcefalias intraútero no se corresponden con los hallazgos posnatales y viceversa, no existe un criterio unánime según los diferentes autores para su diagnóstico y puede ser un cuadro evolutivo a lo largo de la gestación que, por tanto, pase desapercibido hasta el tercer trimestre o incluso hasta el nacimiento17,18.
Cuando el diagnóstico intrauterino es tan complicado, es fundamental el manejo multidisciplinar. El comité de perinatología del hospital facilitó la colaboración estrecha con el servicio de neonatología y neurología infantil, que participaron en el proceso diagnóstico y la toma de decisiones en nuestro caso. La orientación hacia la gravedad del cuadro vino marcada fundamentalmente por tan importante grado de microcefalia que se confirmó de forma posnatal.
ConclusionesEste síndrome es una enfermedad extremadamente rara y carecemos de información acerca del pronóstico y la evolución. Su diagnóstico es de exclusión y la sospecha viene dada por la visualización del linfedema que lleva, tras descartar las principales causas de microcefalia, a realizar un estudio de fondo de ojo que confirma el diagnóstico.
Lo que resulta más complicado es su sospecha antenatal y la explicación a los padres de las consecuencias o secuelas que puede acarrear dicho hallazgo ecográfico, en nuestro caso fundamentalmente la microcefalia marcada y el desarrollo de CIR precoz. Lo importante para el obstetra o perinatólogo que se encuentra ante dicha microcefalia es descartar intraútero las causas principales y valorar el grado de afectación neurológica que puede acarrear.
En este caso, habrá que tener en cuenta el consejo genético de cara a gestaciones posteriores. Dicho síndrome puede tener herencia autosómica dominante, que implica menor retraso mental, herencia autosomia recesiva, que asocia un mayor retraso mental, o tratarse de un cuadro esporádico como parece ser el caso.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.