



INTRODUCCIÓN
La trisomía 22 se considera incompatible con la vida1,2. Se ha descrito en el 8-14% de los casos de abortos espontáneos causados por trisomías autosómicas, y es la segunda trisomía autosómica más frecuente tras la trisomía 163,4. En los años setenta se publicaron los primeros casos de trisomía 22 en neonatos5. En los últimos años se han estudiado algunos casos de trisomía 22 que corresponden citogenéticamente a duplicaciones de la parte distal del cromosoma 11q translocada a la parte proximal del cromosoma 22q6. La trisomía 22 en mosaico sí es compatible con la vida1,7-13. En estos casos se ha comprobado que el cariotipo sanguíneo en linfocitos es normal, pero el realizado en los fibroblastos cultivados procedentes de una muestra cutánea muestra la trisomía 22 en mosaico. Ésta puede estar asociada a un fallo ovárico con fenotipo que recuerda el síndrome de Turner-Ullrich7,9.
Presentamos una paciente de 25 años con talla baja, cúbito valgo, hemiatrofia del hemicuerpo derecho y disgenesia ovárica. La trisomía 22 en mosaico se le detectó en la biopsia de ambas cintillas ováricas, en los fibroblastos cutáneos, pero no en los linfocitos sanguíneos. Se investigó la presencia de la aneuploidía en sus familiares directos, en la creencia de que la disomía parenteral pudiera ser un posible mecanismo de la trisomía 22 en mosaico.
CASO CLÍNICO
Paciente de 25 años, con diagnóstico de amenorrea primaria. Tiene una hermana gemela con menstruaciones regulares. En su infancia se le diagnosticó de enanismo tipo Russell-Silver. En la exploración clínica se apreció una hemiatrofia del hemicuerpo derecho, cúbito valgo y talla baja. Los caracteres sexuales secundarios son propios de la edad excepto el escaso desarrollo mamario. La paciente no debutó con pubertad espontánea y las concentraciones hormonales eran compatibles con los de disfunción ovárica (hormona foliculoestimulante [FSH]: 81 IU, hormona luteinigante [LH]: 22,9 IU, estradiol: 17 pg/ml). Los genitales externos presentaban un aspecto normal. Sin embargo, la exploración de los genitales internos mediante laparoscopia reveló que tanto el útero como las trompas de Falopio tenían una forma irregular y ambos ovarios resultaron ser cintillas blancas largas. La densitometría ovárica diagnosticó una moderada osteopenia. Se le prescribió tratamiento estrógeno-progestágeno para provocar las menstruaciones y restaurar el estatus hormonal.
Estudios citogenéticos
El cariotipo en linfocitos sanguíneos era normal, 46 XX. El que se realizó en las células obtenidas de sendas biopsias ováricas demostró una trisomía 47,XX,+22(7)/46,XX(43) en mosaico en el 14% de las 50 metafases estudiadas (fig. 1). El cariotipo de los fibroblastos cutáneos también mostró 47,XX,+22(18)/46,XX(32), pero en una proporción diferente.
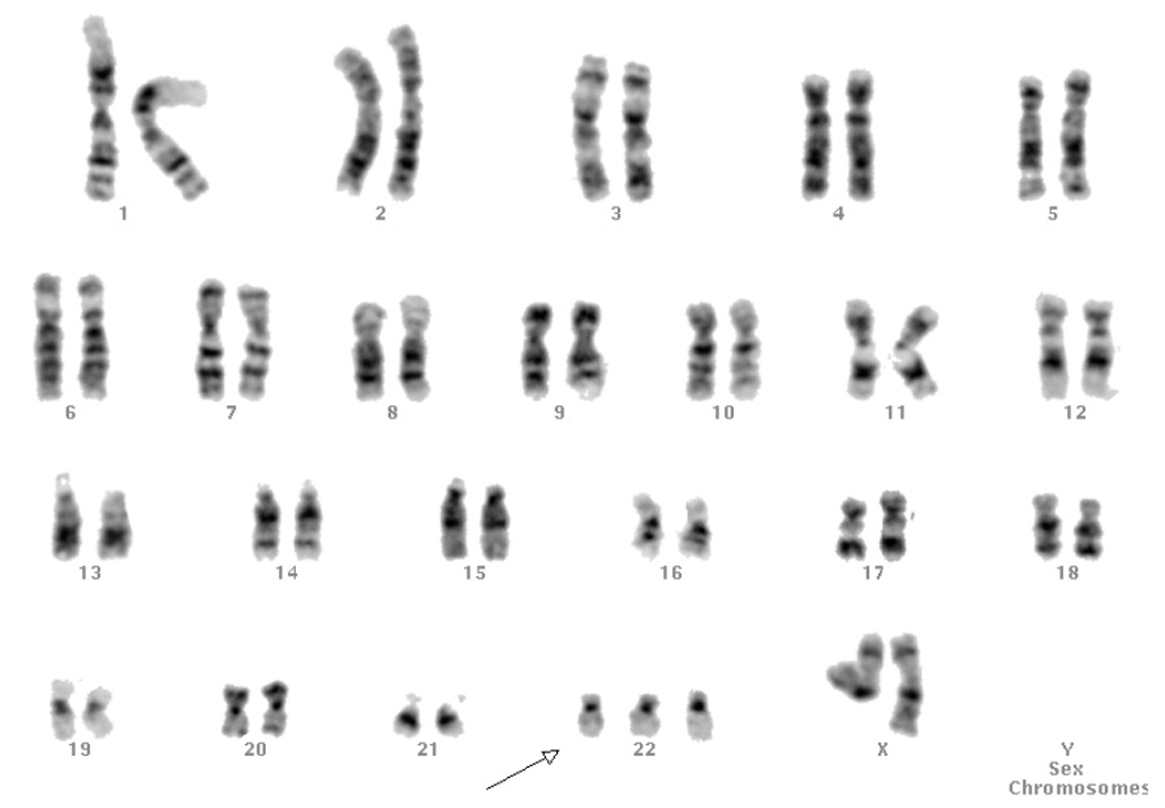
Figura 1.Cariotipo de células de fibrilla ovárica con trisomía 22.
Análisis molecular
Se aislaron muestras de ADN de células sanguíneas y de fibroblastos cutáneos en el caso de la paciente, y ADN de muestras sanguíneas de sus familiares, y en todos los casos se utilizó un procedimiento convencional de análisis "salting out". Se excluyó la presencia del cromosoma Y mediante un grupo de secuencias específicas (sequence tagged sites [STS]) que previamente se amplificaron mediante la reacción en cadena de la polimerasa (PCR), y que se analizaron en gel de agarosa: SY14, sY18, sY68 para el brazo corto, sY78 para la región del centrómero y el análisis de los loci AZFa (SY84, sY85, sY86), AZFb (SY124, sY127, sY134) y AZFc (sY149, sY157) correspondientes al brazo largo del cromosoma Y.
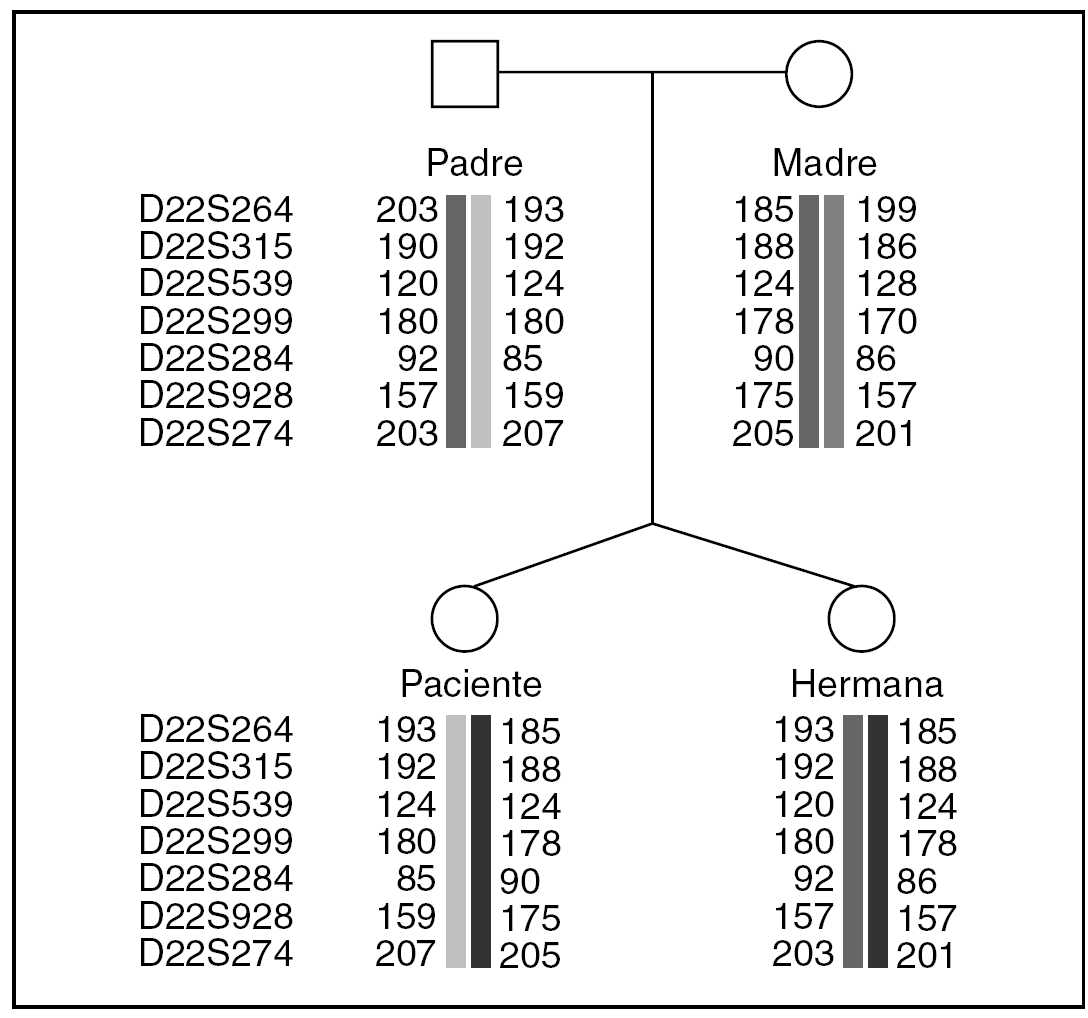
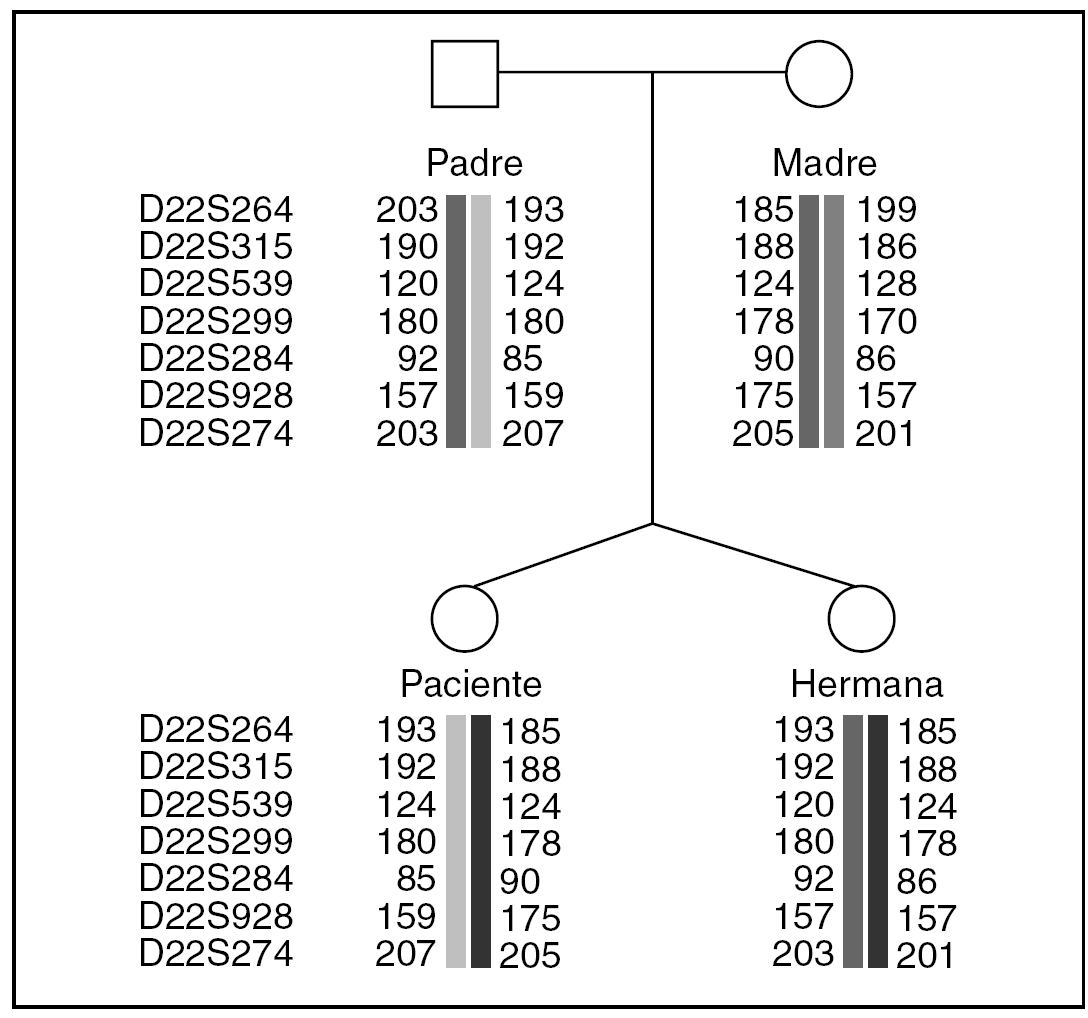
Se excluyó la disomía uniparental mediante "primers" polimórficos microsatélites del cromosoma 22: D22S624, D22S315, D22S539, D22S299, D22S284, D22S928 y D22S274 (Proligo FR®) (fig. 2). Se realizó el análisis de los microsatélites fluorescentes indicados con un analizador genético tipo ABI 310 (Applied Biosystem®).
DISCUSIÓN
El síndrome de Silver-Russel se describió por primera vez en 1953 por Silver et al14, en un bebé que presentaba un peso muy bajo al nacer, talla baja, asimetría corporal y alteraciones en el desarrollo gonadal y gonadotropinas urinarias altas. En una posterior descripción del síndrome, los autores incluyeron los siguientes rasgos físicos: hueso frontal abombado con una cara pequeña triangular; apertura palpebral grande con largas pestañas; boca pequeña con las comisuras desviadas hacia abajo; hipoplasia mandibular; malformaciones dentales; clinodactilia de ambos dedos meñiques, braquidactilia y sindactilia; manchas cutáneas color café con leche, y embarazos que cursaban con gestosis15. La mayoría de los casos de este enanismo son esporádicos, aunque se ha sugerido una posible transmisión hereditaria en algunas familias16. En los embarazos gemelares monocigóticos es posible una presentación discordante, con un feto afectado y el otro normal17. En nuestro caso clínico, la paciente nació de un embarazo gemelar y mostró un retraso de crecimiento intrauterino significativo y una hemiatrofia corporal derecha, mientras que su hermana nació con un peso y aspecto físico adecuados. No se realizaron estudios genéticos en el bebé afectado y se catalogó de enana tipo Silver-Russell, ni tampoco recibió tratamiento alguno para acelerar su desarrollo.
Se han descrito algunas alteraciones cromosómicas en relación con el síndrome de Silver-Russell: 47 XX, UPD (7)mat,+r(7)pat/46,XX18, translocaciones 17;20 y 1;17, deleciones 8q11-q13 y 17q22-q24, deleción del brazo corto del cromosoma 18, trisomía 18 en mosaico o triploidía en mosaico, así como la presencia de la disomía materna en el cromosoma 7 en cerca de 10% de los pacientes19. Se ha identificado un posible gen candidato, GRB10, en una duplicación 7p11.2-p13 que tiene un efecto supresor sobre el crecimiento20. Tanto la trisomía 22 sin mosaico como en mosaico tienen unos rasgos fenotípicos similares a los que se presentan en el síndrome de Silver-Russell21. La hemiatrofia, el retraso mental y el retraso del crecimiento intrauterinos son indicadores inespecíficos de un posible mosaicismo y requieren un estudio citogenético21. Además, muchos signos propios del síndrome de Ullrich-Turner como talla baja, ptosis, cuello alado, cúbito valgo o fallo ovárico, pueden estar presentes en la trisomía 22 en mosaico7,9. La disgenesia ovárica es común en la trisomía 22 en mosaico7,9,11-13 y en otras aberraciones autosómicas citogenéticas2,22. También puede ser el resultado de la duplicación de la banda band 22q12 → qter23. No está claro todavía el papel del cromosoma 22 en la regulación de la función ovárica. Se sabe que ambos cromosomas X deben estar presentes, activos e intactos en los oocitos normales24. El fallo ovárico prematuro comúnmente se produce por la monosomía 45X0, trisomía XXY, mosaicismos 45 X0/46 XX y anomalías estructurales de los cromosomas sexuales (deleciones, inversiones, translocaciones balanceadas, isocromosomas)25-27. En estos casos, los análisis con PCR e hibridación in situ con fluorescencia (FISH) han confirmado la presencia de una "región crítica" en el brazo largo del cromosoma X que debe estar intacto para el mantenimiento de la función ovárica28. Esta región se localiza en Xq13-q26-q2728-31, y loci cercanos de Xp y Xq32. El grado de pérdida de oocitos, atresia y disgenesia ovárica se relaciona con la extensión de la zona afectada dentro de la región crítica, pero no con los genes afectados33. También se ha comunicado fallo ovárico prematuro en mosaicismos de bajo nivel34 y asociado a trisomía 1835. Algunos genes autosómicos pueden estar relacionados con la diferenciación ovárica y la disgenesia32,36. Las pacientes con disgenesia ovárica pueden o no presentar características del síndrome de Ullrich-Turner30. En nuestro paciente, el cariotipo sanguíneo era 46 XX y no se demostró presencia alguna de mosaicismo o del cromosoma Y. La laparoscopia reveló unos genitales internos anormales y cintillas en vez de ovarios. La biopsia de dichas cintillas para su estudio citogenético confirmó una trisomía 22 en mosaico que hubiese quedado sin diagnosticar si no se hubiera realizado una laparoscopia. El diagnóstico genético final puede también lograrse con el estudio de los fibroblastos obtenidos de una muestra de piel, sobre todo en casos de pacientes con disgenesia ovárica y signos del síndrome de Ullrich-Turner y asimetría corporal9. Se excluyó la presencia de material del cromosoma Y en las muestras gonadales. El análisis segregacional familiar no confirmó la presencia de una disomía parental que afectase el cromosoma 22. Somos conscientes de que los resultados de la biopsia cutánea y las muestras de ADN sanguíneo pueden explicarse por un mosaicimo de bajo grado; no obstante, creemos que los resultados de nuestra paciente se deben a un error mitótico posfertilización como posible mecanismo que explicase la presencia de la trisomía 22 en mosaico en la paciente y su ausencia en sus padres y hermana37.
La distribución del mosaicismo en el concepto depende del momento, la línea celular afectada, la viabilidad celular y el cromosoma involucrado37. El desarrollo consecuente del mosaicismo está relacionado con su tipo meiótico o somático.
El mosaicismo meiótico a menudo se asocia con un efecto adverso más severo en el concepto debido a la presencia de disomía uniparental en el embrión/feto y/o la disfunción de una placenta trisómica37. Como el mosaicismo puede ser específico del tejido, un resultado normal del cariotipo en linfocitos sanguíneos cultivados no excluye la presencia del mosaicismo en cualquier parte del concepto37. En este caso, el mosaicismo puede detectarse mediante la combinación de un análisis citogenético y con técnicas moleculares como FISH y la hibridación genómica comparativa37. La disomía uniparental consiste en la falta de disyunción de una pareja de cromosomas en la meiosis, por lo que un gameto tiene 24 crosomosomas. La disomía uniparental materna (DUPM) es más frecuente que la paterna (DUPP) en una proporción 3:1, y es responsable de la aparición de impresión genómica aberrante, homocigosidad de mutaciones que afectan a genes autosómicos recesivos, homocigosidad de desórdenes del cromosoma X en mujeres y transmisión de padre a hijo de rasgos asociados al X, algunas veces con evolución desfavorable38. En ocasiones, después de la fertilización de un gameto disómico por un gameto monosómico para el mismo cromosoma, se produce la pérdida consecuente del cromosoma normalmente heredado ("rescate trisómico"), dando lugar al mosaicismo en la placenta o en tejidos fetales38,39. Este mosaicismo de bajo nivel puede permanecer oculto si sólo se realiza el cariotipo en linfocitos sanguíneos pero no en fibroblastos cutáneos u otros tejidos específicos38,39, como así ha ocurrido en la paciente. Se ha descrito el retraso de crecimiento intrauterino fetal severo asociado a trisomía 22 con y sin mosaico en la placenta y con disomía uniparental con un contingente cromosómico normal del feto40,41. El origen paterno o materno del cromosoma extra no parece tener importancia en la supervivencia posterior de la trisomía 2242.
AGRADECIMIENTOS
Expresamos nuestro agradecimiento a la Sra. María José Bernabé Espinosa por la asistencia técnica en la preparación de las muestras de ADN y los estudios del cromosoma Y, a la Sra. Piedad Salas por la asistencia técnica en la FISH y a la Sra. María Carmen Bernabé Martínez por la asistencia técnica en la preparación de los cromosomas de las células cultivadas.